Genesis Workbench: Um projeto para aplicativos de ciências da vida em Databricks
por Peter Hawkins, May Merkle-Tan, Srijit Chandrashekhar Nair, Eli Swanson, Yang Yang, Guanyu Chen, Ram Goli e Douglas Moore
A AI generativa está transformando a P&D em biotecnologia ao utilizar modelos de base treinados em diversos datasets biológicos para viabilizar aplicações avançadas na descoberta de medicamentos, modelagem preditiva e medicina personalizada. No entanto, os cientistas da área muitas vezes enfrentam barreiras técnicas e administrativas significativas, que podem desviar a atenção das principais atividades de pesquisa. Isso inclui: configurar ambientes de GPU, gerenciar fluxos de trabalho complexos e garantir controles de acesso apropriados. O Genesis Workbench é um esforço para enfrentar esses desafios, fornecendo um blueprint para o desenvolvimento de aplicações de ciências da vida usando modelos de base para biologia na Databricks Platform.
A AI está acelerando a descoberta de alvos e transformando o design de medicamentos
As ciências da vida estão passando por uma transformações fundamental, impulsionada pela convergência de enormes dataset genômicos e multi-ômicos com avanços em AI (AI) e machine learning (ML). As ferramentas de AI, inicialmente especializadas, evoluíram rapidamente para modelos de base sofisticados e generalizados, demonstrados por inovações na previsão de estruturas de proteínas, modelos de linguagem de proteínas e até mesmo modelos generativos avançados. Esses modelos básicos, treinados em um vasto corpus de dados estruturais e de sequências biológicas, estão permitindo a descoberta acelerada de medicamentos por meio de processos como o design lab-in-the-loop, melhorando a eficiência das tarefas diárias dos cientistas e, possivelmente, facilitando o design de novo. Essa aceleração é apoiada ainda mais por modelos especializados de linguagem em larga escala (LLMs), que são essenciais para a extração e a síntese precisas do conhecimento biomédico da literatura científica, dos Registros Eletrônicos de Saúde (EHRs) e dos Notebooks Eletrônicos de Laboratório (ELNs).
Desafios práticos na construção de uma plataforma de pesquisa segura e escalável
As tecnologias para encontrar alvos ou desenvolver novos medicamentos com mais eficiência, bem como os dados usados nelas, são Propriedade Intelectual (IP) altamente protegida. Portanto, as organizações precisam garantir que controles de acesso apropriados possam ser concedidos a indivíduos e grupos, tanto para os dados quanto para os modelos. Outras questões de governança, como a auditoria do uso de modelos e o monitoramento de custos, são key fatores para modelos de base em dataset biológicos de grande escala. No entanto, é importante que esses processos de controle de acesso não sejam tão difíceis de implementar ou tão opacos para os usuários a ponto de impedir o progresso real da comunidade científica dentro da organização. Na prática, muitas organizações têm dificuldade em encontrar esse equilíbrio.

Apesar de sua experiência em biologia ou biologia computacional, muitos cientistas altamente talentosos têm dificuldade para configurar modelos biológicos avançados devido à carga de tarefas relacionadas a aspectos de nicho da moderna tecnología de AI. Esses desafios incluem complexidades técnicas, como a configuração de ambientes CUDA para aceleração de GPU, o que é essencial para o treinamento eficiente de modelos grandes. Além disso, os cientistas geralmente precisam criar e gerenciar fluxos de trabalho complexos que automatizam e dimensionam com eficiência o processamento de dados, o treinamento de modelos e os MLOps. Essas tarefas geralmente exigem habilidades fora do treinamento biológico tradicional. A engenharia de dados também representa um obstáculo significativo, pois envolve a coleta, a limpeza e a integração de diversos datasets biológicos e, ao mesmo tempo, garante o compliance com as políticas de governança de dados para manter a privacidade e a reprodutibilidade. Essas demandas não biológicas desviam tempo e foco valiosos da pesquisa científica principal, retardando o progresso e a inovação na aplicação de modelos de AI generativa nas ciências da vida. Para resolver essa lacuna, é necessário haver colaboração interdisciplinar e melhor acesso a ferramentas que reduzam as barreiras técnicas para os pesquisadores biológicos.
Genesis Workbench: um modelo para AI/ML biológica no Databricks
Poder sem esforço: o Databricks simplifica os dados e a AI
O Databricks se destaca como uma plataforma de análise unificada que combina governança robusta, usabilidade intuitiva e recursos abrangentes para criar qualquer solução de dados ou AI que você precisar. Com ferramentas centralizadas para gestão de dados, segurança e compliance, ele garante que seus dados estejam sempre protegidos e, ao mesmo tempo, permaneçam facilmente acessíveis a usuários de todos os níveis de habilidade. Seus recursos de colaboração contínua, potência de processamento dimensionável e suporte para todo o espectro de cargas de trabalho de análise de dados e AI fazem dele a base ideal para organizações que buscam inovar e, ao mesmo tempo, manter controle e simplicidade rigorosos. Organizações grandes e pequenas obtiveram sucesso na criação de modelos biológicos na Databricks, desde a família TEDDY de modelos de fundação da Merck até o Atlas de célula única da Tahoe Therapeutics.
O Genesis Workbench ajuda você a potencializar as ciências da vida no Databricks

O Genesis Workbench fornece um modelo para o desenvolvimento de aplicativos de ciências da vida que aproveitam os recursos do Databricks. Ele fornece padrões de trabalho que utilizam recursos como fluxos de trabalho automatizados, clusters de GPU, servindo modelo e MLflow para acelerar a pesquisa em ciências da vida orientada por AI. Ele apresenta uma interface intuitiva do Databricks Apps com modelos biológicos pré-empacotados e fluxos de trabalho personalizados, permitindo que os cientistas comecem rapidamente sem configurações complexas.
Em colaboração com a NVIDIA, o BioNeMo—uma estrutura de AI generativa para biologia digital—está integrado para facilitar o acesso a modelos avançados pré-treinados. Os modelos BioNeMo são otimizados para o hardware NVIDIA, oferecendo alto desempenho e escalabilidade para cargas de trabalho corporativas.

Por ser de código aberto, o Genesis Workbench oferece padrões extensíveis para engenheiros de AI, reduzindo a carga de trabalho não biológica e promovendo a inovação rápida ao usar e combinar modelos de AI de base para biologia. Oferecemos uma variedade de modelos com o Genesis Workbench como ponto de partida, que podem ser usados tanto por cientistas de bancada pelo aplicativo quanto por usuários computacionais avançados que criam pipelines. Um ponto importante é que, ao servir modelos em APIs, você abstrai as dependências complexas do modelo e os requisitos de GPU. Isso permite que os usuários integrem ferramentas de uso comum, mas muito complexas, em pipelines únicos.
O Genesis workbench é um Solution Accelerator da Databricks que está em desenvolvimento ativo, e esperamos que nosso conjunto de recursos continue crescendo.
Arquitetura do Genesis Workbench
Este aplicativo foi desenvolvido na plataforma Databricks e usa os seguintes recursos da plataforma
- Unity Catalog para governança
- Aplicativos Databricks para UI
- GPU Model Serving para servir modelos de fundação
- Lakeflow Jobs para executar inferência em lote dimensionável e ajuste fino
- Compute clássico com GPUs para cargas de trabalho interativas e em lotes
- Docker Container Services para integração de bibliotecas de terceiros
- Databricks ativo Bundle para facilitar a implementação
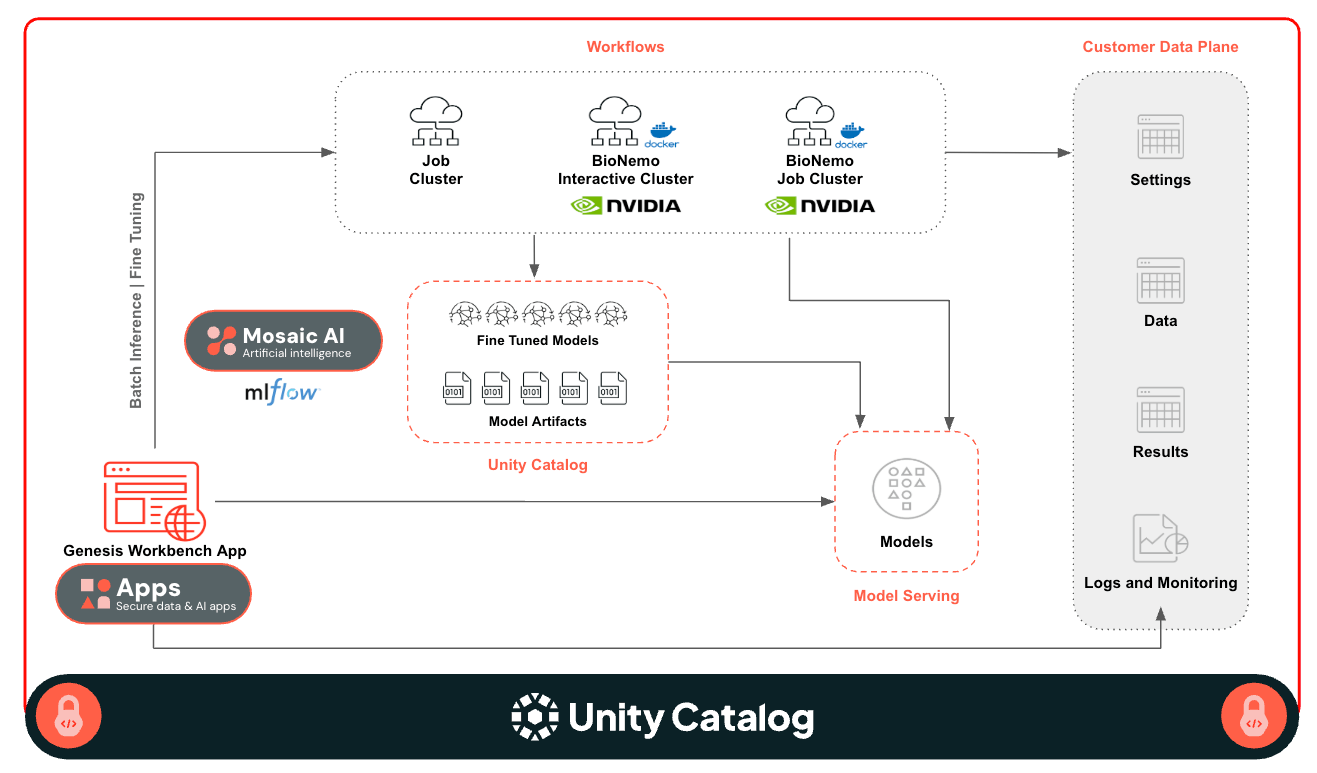
Módulos no Genesis Workbench
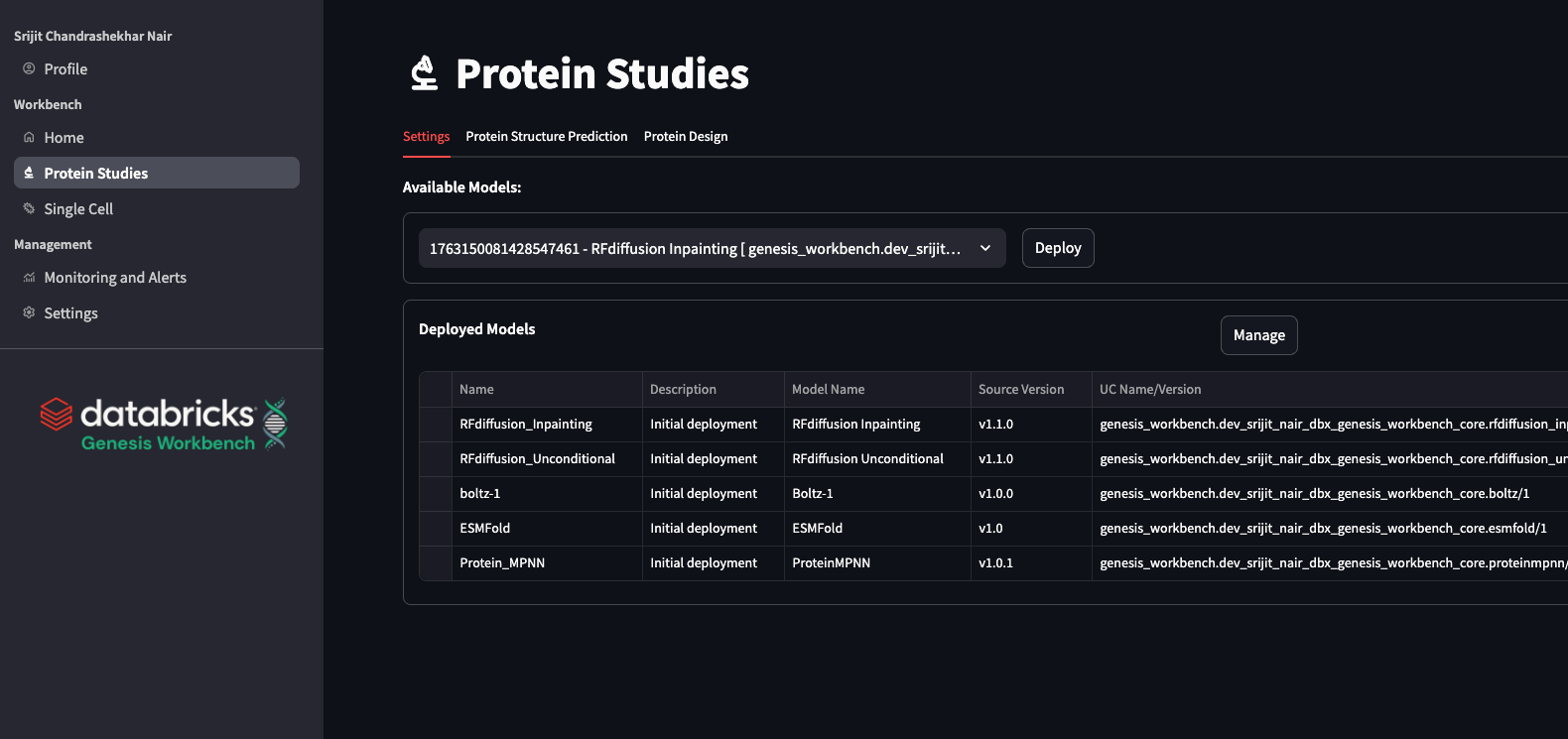
Enovelamento e projeto de proteínas
Visão geral
A capacidade de prever computacionalmente a estrutura tridimensional de uma proteína a partir de sua sequência de aminoácidos tem sido um problema computacional e teórico de longa data. A competição CASP tem sido um local onde os avanços mais recentes são testados uns contra os outros, promovendo as capacidades de modelagem da comunidade de pesquisa. A DeepMind, com seu modelo inicial alphafold (Senior et al. 2020, Nature) e, posteriormente, com os lançamentos do alphafold2 (Jumper et al. 2021, Nature) na competição CASP, conseguiu melhorar substancialmente o desempenho da previsão de estrutura. Para isso, eles utilizaram técnicas de AI para aprender com estruturas medidas anteriormente e incorporar informação de grandes bancos de dados de sequências. A capacidade de prever muitas estruturas com precisão quase experimental, incluindo multímeros de proteínas, revolucionou a abordagem da descoberta de medicamentos.
Agora existe uma grande variedade de modelos para tarefas relacionadas à estrutura de proteínas. Modelos como o Alphafold-3 (Abramsom et al. 2024 Nature) (pesos fechados), Boltz-1 (Wohlend 2024, BioRxiv) e Chai-1 (Chai discovery team 2024, BioRxiv) agora vão além das proteínas e abrangem estruturas mais avançadas que envolvem proteínas, DNA, RNA e moléculas pequenas. Outros modelos abertos, como o Openfold3(equipe do Openfold3), foram lançados recentemente com recursos semelhantes, e espera-se que esse zoológico de modelos continue crescendo. Além disso, a capacidade de gerar proteínas com modelos generativos está avançando rapidamente; ferramentas como RFdiffusion (Watson et al. 2023, Nature) e ProteinMPNN (Dauparas et al. 2022, Science) são usadas com frequência nesse espaço (consulte, por exemplo Bielska et al. 2025, front.immunol.), e novos modelos, como o BoltzGen (Stark et al. preprint, repo) e outros, fornecem um conjunto de ferramentas em rápida evolução para a descoberta e o design computacional de medicamentos
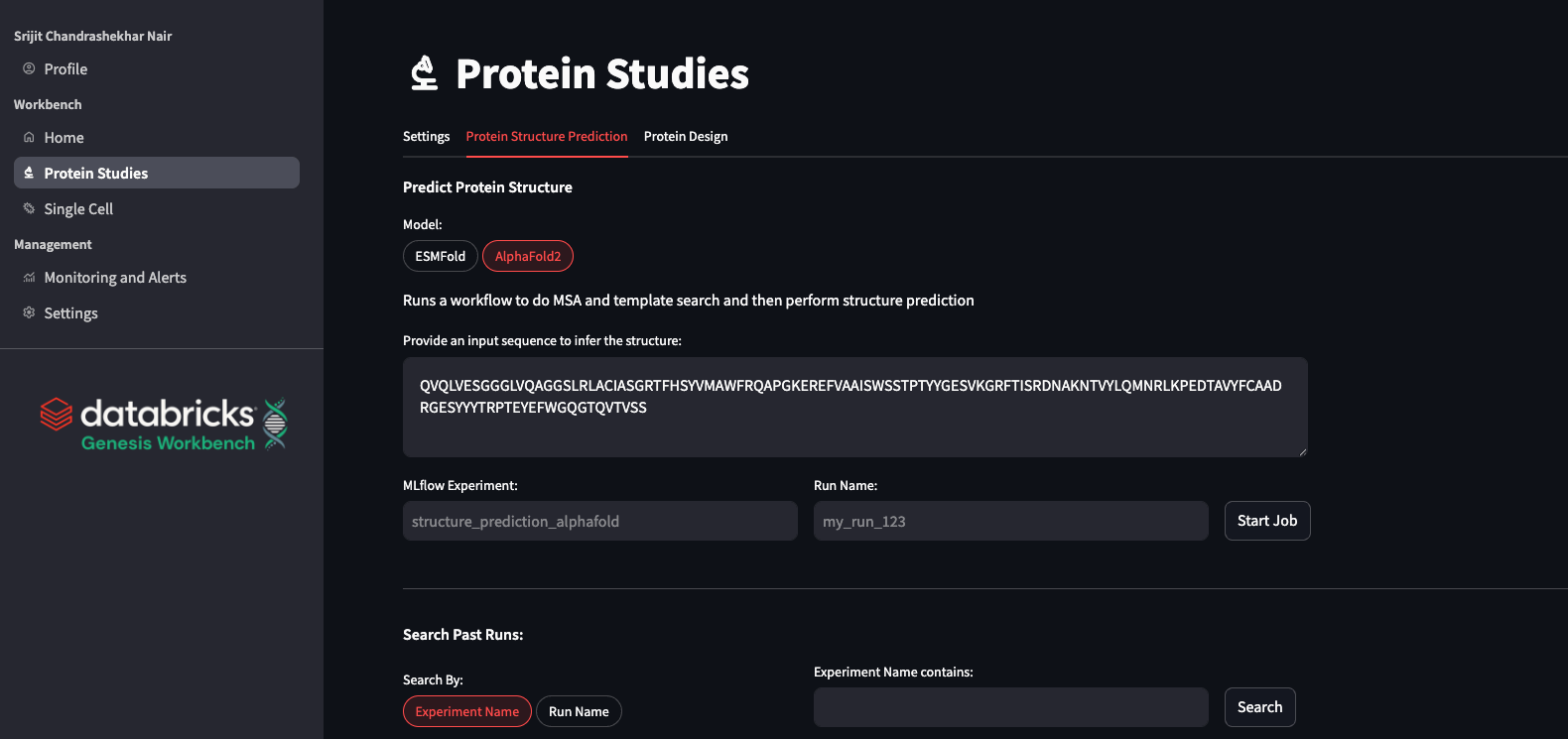
Alphafold
Incluímos a versão estável mais recente (v2.3.2) do alphafold2 (Jumper et al. 2021, Nature). Como o processo de alphafold leva algum tempo para ser executado devido à extensa computação de recurso, no Genesis Workbench, mostramos a você como provisionar o alphafold como um Job de fluxo de trabalho no Databricks. O job é composto de duas tarefas: a tarefa dominada pela CPU (MSA, extração de recurso) e a tarefa de dobragem dominada pela GPU. Isso nos permite execução de cada tarefa no tipo de compute apropriado. O job utiliza um sistema de filas, muito parecido com um HPC, permitindo que várias sequências sejam dobradas simultaneamente enquanto outras aguardam na fila.
ESMFold
O ESMFold (Lin et al. 2023, Science) é um modelo de aprendizagem profunda rápido e econômico para a previsão da estrutura de proteínas que utiliza modelos avançados de linguagem de proteínas do ESM e um cabeçalho estrutural para inferir estruturas de proteínas a partir de uma única sequência. Embora sua precisão seja menor do que a dos modelos que usam alinhamentos de várias sequências, o ESMFold é suficiente para muitas aplicações práticas. No Genesis Workbench, implantamos o ESMfold, envolvido em um modelo MLflow, e o registro no Unity Catalog. Servir um modelo MLflow registrado no Unity Catalog como uma API é extremamente simples no Databricks, incluindo suporte para operações de escala até zero para economia de custos. Com o ESMFold servido como uma API, os pesquisadores podem prever novas estruturas de proteínas rapidamente, integrá-lo facilmente a pipelines computacionais e visualizar ou fazer download dos resultados para análise posterior usando padrões mostrados no Genesis Workbench.
Boltz
O Boltz-1 (Wohlend 2024, BioRxiv) oferece um modelo de código aberto e pesos (MIT) para previsão de estrutura biomolecular. Assim como o ESMFold, fazemos o registro do Boltz-1 como um modelo no Unity Catalog e o servimos em um endpoint de model serving. O Boltz-1 tem uma entrada MSA opcional; é possível omiti-la, usar um MSA pré-calculado ou usar um endereço de servidor mmseqs2 (Steinegger, Sölding 2017 Nat.Biotech). Para garantir uma variedade de opções para o Boltz-1, encapsulamos os componentes essenciais do modelo Boltz-1 como um pacote Python junto com as opções do JackHMMer (Johnson et al. 2010 Bioinformatics) da biblioteca alphafold2 (Jumper et al. 2021, Nature). Isso permite que os usuários escolham o JackHMMer MSA, o que pode ser vantajoso para alguns, já que o endereço do servidor mmseqs2 default é um URL público, o que viola a maioria das políticas de segurança.
As funções do Boltz-1 e os estágios do MSA são encapsulados com o mlflow tracing. O rastreamento permite visualizar facilmente todos os estágios da inferência, além da entrada e saída de cada estágio, como, por exemplo, o estágio do MSA. Isso pode ser especialmente útil ao usar o modelo em um Notebooks, pois permite ver rapidamente o que aconteceu no modelo e identificar problemas com o MSA sem precisar navegar para arquivos armazenados separadamente.
Design de proteínas com ProteinMPNN e RFDiffusion
O design computacional de proteínas é um espaço em rápida evolução com grande potencial para mudar a forma como projetamos proteínas eficazes tanto para a atividade enzimática quanto para os efeitos terapêuticos. O RFdiffusion (Watson et al. 2023, Nature) e o ProteinMPNN (Dauparas et al. 2022, Science) são frequentemente usados juntos no design de proteínas, por exemplo, no design computacional de anticorpos (Bielska et al. 2025, front.immunol.) e, recentemente, até mesmo para o design de anticorpos de novo (Bennett et al. 2025, Nature).
No Genesis Workbench, demonstramos como disponibilizar o RFDiffusion e o ProteinMPNN. Demonstramos como, uma vez que as dependências complexas são abstraídas no servindo modelo, é fácil unir esses modelos em pipeline computacionais. Em particular, combinamos ESMFold, RFdiffusion e ProteinMPNN e chamamos todo o pipeline de uma pequena máquina somente com CPU que hospeda o aplicativo Genesis Workbench. Não precisamos mais manter todas as dependências de cada modelo em um único local ou nos preocupar com versões conflitantes do CUDA. No aplicativo, também alinhamos estruturalmente as saídas com a estrutura original prevista para exibir as sequências originais e as projetadas no visualizador Mol*.
Isso demonstra como os usuários podem criar novas ferramentas com base nos componentes do Genesis Workbench para criar processos personalizados para os objetivos de sua organização ou equipe de pesquisa.
Análise de célula única
Visão geral
A transcriptômica de célula única é uma tecnología avançada para entender as populações de células em diferentes coortes, identificar tipos de células de nicho, elucidar trajetórias de células e muito mais. A escala de dados disponíveis nesse espaço está crescendo rapidamente, não apenas pelo aumento do processamento de amostras, mas também pelo aprimoramento das tecnologías. Isso representa um desafio para a rotulagem e o processamento de dados nessa escala, levantando questões:
- Alguma parte da análise pode ser automatizada para evitar sobrecarregar os pesquisadores avançados de nível de doutorado, exigindo que eles dediquem tempo a tarefas repetitivas?
- Como podemos pré-anotar os dados para aprimorar o descobrimento de dados para futuras análises em nível de atlas das amostras?
Nosso objetivo é fornecer soluções para esses desafios e usar como base nosso blueprint para adaptar essas abordagens a variações desses problemas.
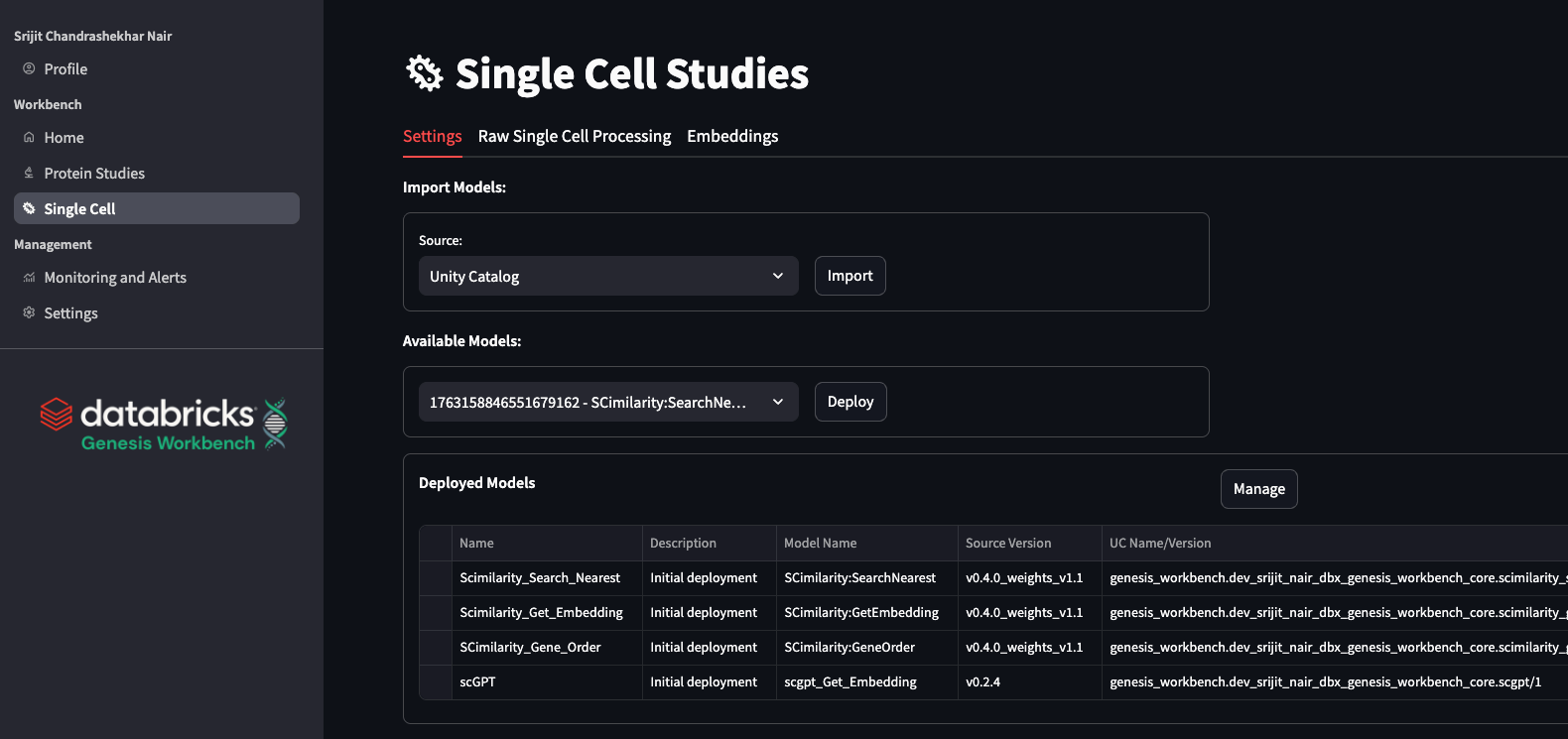
No Genesis Workbench, fornecemos ferramentas para automatizar o processamento de uma única célula e visualizar os resultados, todos rastreados em experimentos MLflow, para que os usuários mantenham o controle sobre os dados e como eles são compartilhados com seus colegas. Isso inclui pipelines de CPU com seleção ideal de RAM e fluxos de trabalho acelerados por GPU. Além disso, incluímos visualizações de dados responsivas e de baixa latência dos dados processados. Também incorporamos modelos básicos de expressão de célula única: SCimilarity (Heimberg et al., 2024, Nature) e scGPT (Cui et al., 2024, Nature Methods); para anotação, incorporação e pesquisa em nível de atlas.
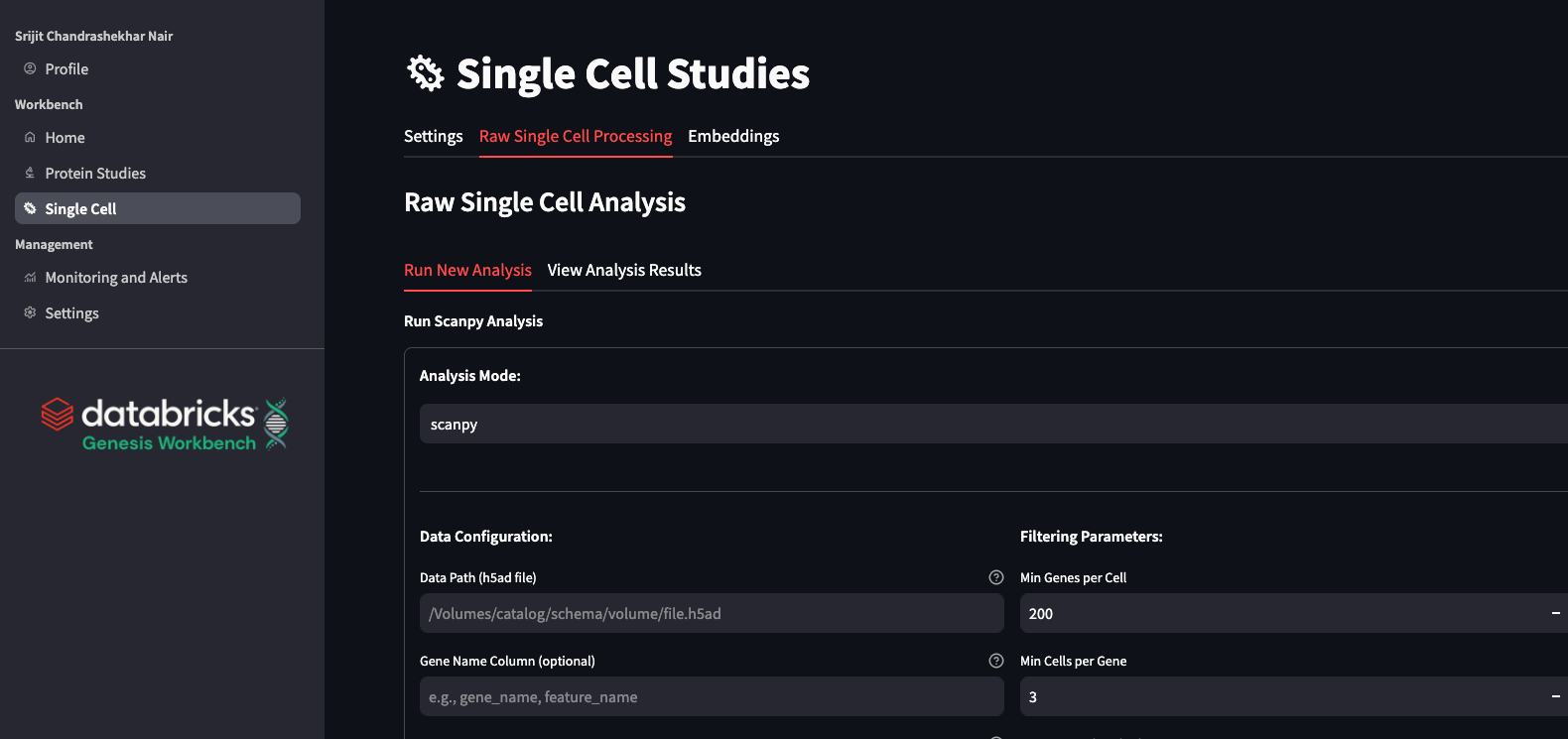
Processamento de dados de scRNA: scanpy e rapids-singlecell
No Genesis Workbench, construímos um job de fluxo de trabalho para uma execução padrão do scanpy (Wolf et al. 2018 Genome Biol), que tenta selecionar automaticamente o menor tamanho de RAM para reduzir a carga do usuário e, ao mesmo tempo, reduzir o custo. O Job logs os parâmetros escolhidos, bem como várias métricas, resultados e números em uma única execução do MLflow. Isso permite um procedimento moderno de acompanhamento de experimentos, com registro automatizado de todos os detalhes relacionados ao pipeline que está sendo executado. Além desse job do Scanpy, também fornecemos um job do RAPIDS-singlecell (Dicks et al., 2022, repo, doi). Esse pacote se comporta de forma muito semelhante ao scanpy e tem as mesmas opções de parâmetros, mas é acelerado por uma GPU. Isso permite um throughput mais rápido, o que é essencial para determinados projetos.
No aplicativo Genesis Workbench, tornamos esses pipelines facilmente acessíveis aos usuários por meio de uma interface simples que permite começar execuções com diferentes parâmetros e view o resultado no aplicativo. Realizamos a curadoria inteligente de dados para permitir uma análise rápida dos resultados no aplicativo, oferecendo suporte a vários usuários em um único compute de aplicativo compacto. Detalhes como métricas de CQ, clusters e expressão de genes marcadores podem ser todos visualizados no aplicativo. Esse processamento pode ser um passo importante antes de você usar dados com modelos de base para scRNA.
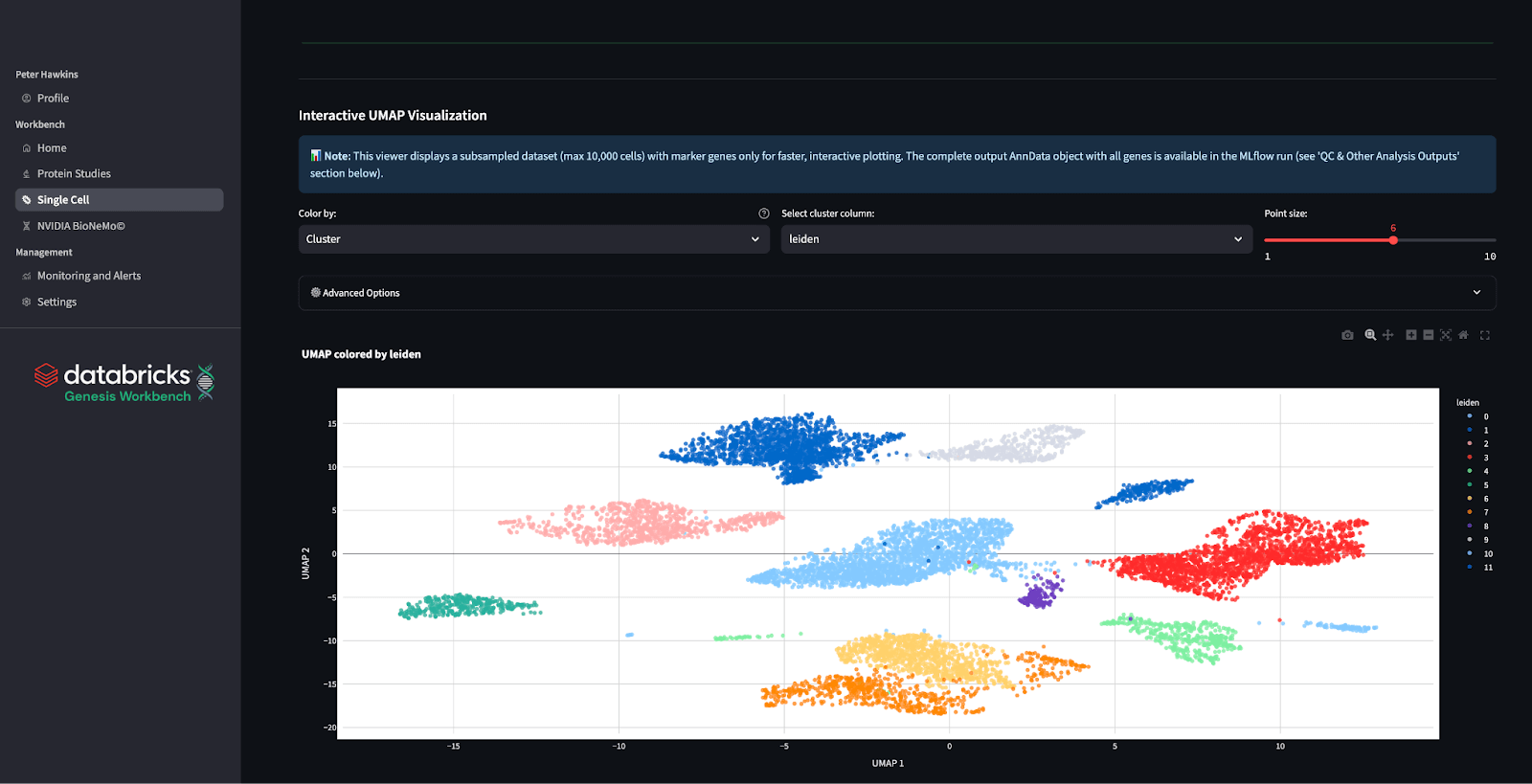
Modelos básicos: SCimilaridade e scGPT
O módulo de análise de célula única do Genesis Workbench oferece aos pesquisadores de ciências biológicas acesso a dois modelos de base de última geração: SCimilarity e scGPT.
O SCimilarity utiliza o aprendizado de métricas para pesquisar em um atlas de células em grande escala para a identificação rápida de populações relevantes para doenças e modelos in vitro análogos, alcançando precisão no nível de órgãos e doenças usando catálogos de referência sem a necessidade de clusterização. Como complemento, o scGPT emprega uma arquitetura de transformador de 53 milhões de parâmetros com incorporação dupla de genes e células para previsão de perturbações, inferência de rede regulatória e integração de multiômica de disparo zero. Juntos, eles harmonizam diversos datasets, minimizam o viés de anotação e revelam percepções mecanicistas sobre doenças - similaridade por meio de gradientes integrados e scGPT por meio de mecanismos de atenção - impulsionando a descoberta translacional e a inovação terapêutica em pesquisas sobre oncologia, imunologia e doenças raras (Heimberg et al., 2024, Nature; Cui et al., 2024, Nature Methods).
O Genesis Workbench implementa os principais pontos de extremidade para ambos os modelos, empacotando esses modelos básicos com clusters de GPU pré-configurados, fluxos de trabalho automatizados e integração com o MLflow: cell_query.gene_order, cell_embedding.get_embeddings, e cell_query.search_nearest para SCimilarity; inferência de incorporação de genes para scGPT. O Notebook de exemplo demonstra o fluxo de trabalho com base no tutorial de miofibroblastos de IPF da SCimilarity — carregamento e normalização de dados de scRNA-seq, computação de embeddings e consulta de 23,4 milhões de células para identificar as populações mais próximas enriquecidas pela doença usando os Endpoints fornecidos. As populações identificadas permitirão a análise scGPT downstream para inferência regulatória de genes, triagem de perturbação e previsões de CRISPR em um único ambiente.
A Databricks e a NVIDIA oferecem velocidade e escala
A pilha de software da NVIDIA oferece soluções poderosas para pesquisas aceleradas em ciências biológicas. O NVIDIA BioNeMo é uma estrutura de código aberto que acelera o desenvolvimento de modelos de aprendizagem profunda para a indústria biofarmacêutica. Ele permite que os pesquisadores escalem os modelos de AI biomolecular, abrangendo dados de DNA, RNA e proteínas, para novos patamares com ferramentas rápidas e simplificadas. O Parabricks oferece análise genômica rápida e de alto throughput usando algoritmos otimizados por GPU, permitindo o processamento rápido de dados de sequenciamento de última geração para laboratórios de genomics. O Rapids-SingleCell aprimora a análise de dados de célula única, permitindo a exploração e a análise escalonáveis e interativas de grandes datasets multiômicos, aproveitando a aceleração da GPU para obter percepções biológicas mais profundas.
A estreita colaboração entre as equipes de engenharia da NVIDIA e da Databricks permitiu a integração perfeita da pilha de software avançada da NVIDIA no Genesis Workbench, fornecendo fluxos de trabalho rápidos e escalonáveis alimentados por AI para ciências biológicas. Usando padrões no Genesis Workbench, você pode:
- Aproveite os modelos BioNeMo no compute de GPU escalável dentro da Databricks Platform
- Faça o ajuste fino de modelos disponíveis no pacote BioNeMo e use-os para inferência em grande escala
- Use o RAPIDS-singlecell, uma estrutura de código aberto acelerada por GPU para análise de célula única
- Use o parabricks para análise genomics
Visibilidade Simplificada e Otimização de Custos com os Dashboards do Databricks
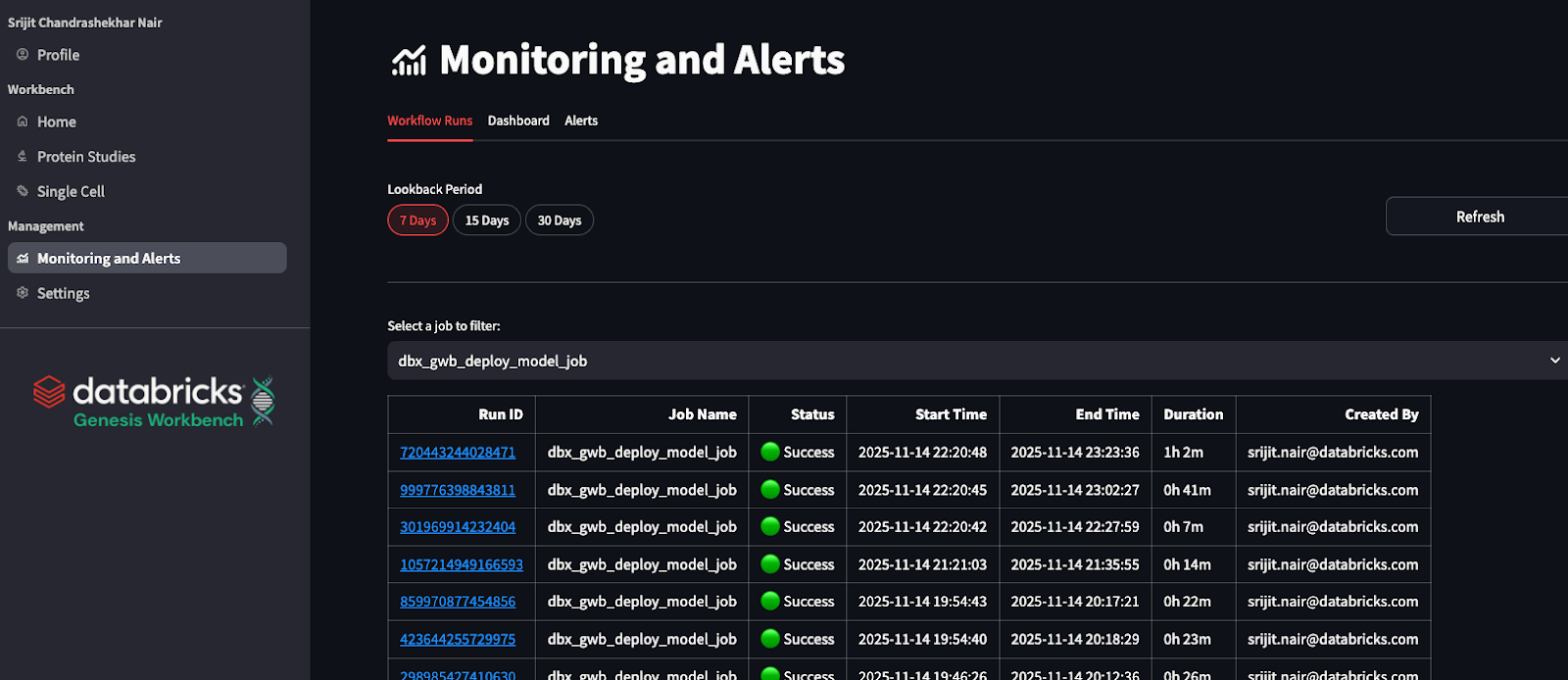
Monitoramento de execuções de Job
Poucos fluxos de trabalho exigem processos de segundo plano de longa duração, e o aplicativo utiliza o Databricks fluxo de trabalho e inicia Jobs de forma assíncrona. O Genesis Workbench inclui um dashboard de monitoramento dedicado que exibe todos os Jobs do Databricks fluxo de trabalho iniciados no sistema.
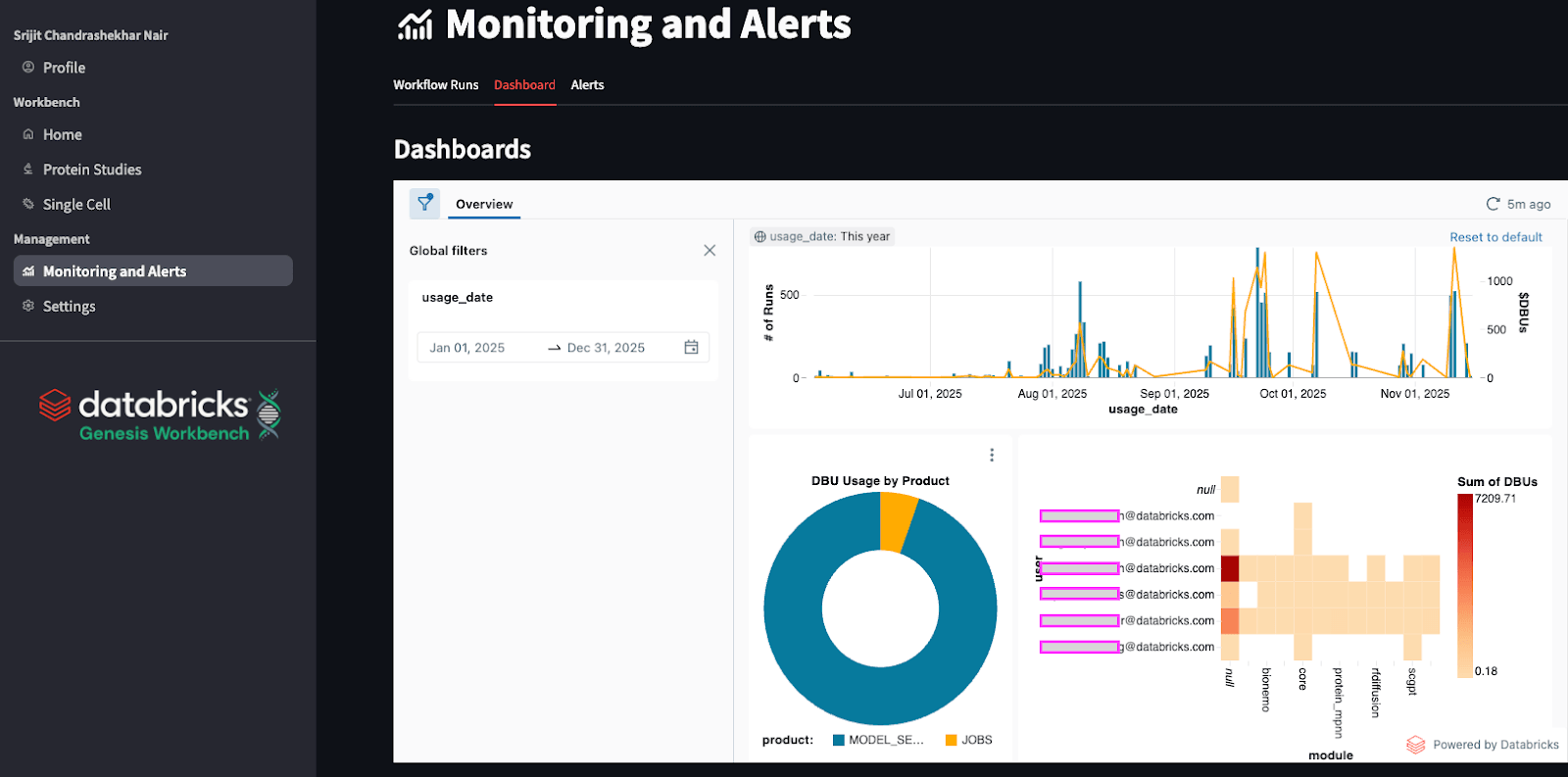
Monitoramento de Custos
A criação de pipelines e ferramentas computacionais com modelos de fundação geralmente levanta questões sobre os custos associados. O Databricks captura informações sobre o uso e o faturamento automaticamente nas tabelas do sistema. Isso significa que você pode criar facilmente painéis sobre essa informação para criar maneiras personalizadas de view e analisar rapidamente o uso e os gastos. Você também pode usá-los para criar alertas automáticos relacionados ao uso excessivo. No Genesis Workbench, fornecemos automaticamente um painel dentro do aplicativo que mostra as despesas em cada modelo, divididas por tempo e usuário. Além disso, o formato do painel também é de código aberto e pode ser personalizado pelas equipes que instalam o Genesis Workbench.
Passos seguintes
Instalação
O Genesis Workbench inclui um script que utiliza os Databricks Asset Bundles para implantar o aplicativo. Você pode fazer o download e instalar o aplicativo no repository do GitHub seguindo as instruções de instalação fornecidas.
A fonte contém:
- scripts para implantar o módulo central do Genesis Workbench em seu workspace que inclui o aplicativo de interface do usuário
- para implementar os módulos abaixo:
- Módulo de célula única
- Módulo de estudos de proteínas
- Módulo BioNeMo que contém definições de contêineres e fluxos de trabalho (OBSERVAÇÃO: o contêiner BioNeMo precisa ser criado separadamente)
- Monitoramento e painéis de controle
Feedback
Este projeto está atualmente em desenvolvimento ativo, e estamos ansiosos para receber seu feedback. Entre em contato com sua equipe de account para discutir seu caso de uso e entender como você pode aproveitar os padrões discutidos no Genesis Workbench.
Mapa rodoviário
- Mais modelos disponíveis prontos para uso
- Suporte avançado a MSA para enovelamento de proteínas mais rápido com MSAs
- Suporte de transmissão para dados de célula única
- Integração do BioNemo com servindo modelo
- Suporte para transcriptômica espacial
Conclusões
O Genesis Workbench oferece modelos de base biológica de MLflow em pacote prontos para uso e modelos NVIDIA BioNemo, que se integram perfeitamente ao ecossistema da Databricks. Este é um modelo de como os usuários do Databricks podem trazer novos modelos para o Databricks, combinar vários modelos em pipelines complexos e dar suporte a cientistas de bancada e computacionais com uma única estrutura.
A governança da Databricks por meio do Unity Catalog oferece um sistema consistente e abrangente para controlar o acesso a modelos, Jobs, dados e ao aplicativo. Isso pode simplificar drasticamente o desenvolvimento de aplicativos e pipelines de dados usando esses modelos de base e, ao mesmo tempo, garantir o controle de acesso adequado.
Os recursos de servindo modelo do Databricks permitem que cientistas computacionais e engenheiros de MLOps abstraiam as dependências complexas e os requisitos de GPU de cada modelo. Isso permite que outros usuários computacionais criem pipelines, integrando múltiplos modelos com suas próprias dependências, sem precisar se preocupar com isso. Isso acelera drasticamente o ciclo de vida de desenvolvimento e pode ser facilmente compartilhado entre equipes com as permissões adequadas, graças ao Unity Catalog.
O Databricks Apps oferece um ciclo de vida de desenvolvedor rápido para testar e criar aplicativos com base nesses blocos de construção de modelos básicos e visualizadores de dados biológicos. Como o código do Genesis Workbench é de código aberto, as equipes podem adaptar e refinar o aplicativo para atender às necessidades de suas equipes. Eles podem adicionar novos modelos conforme necessário, bem como seguir o modelo fornecido pelo Genesis Workbench para criar o sistema conforme necessário para seus negócios.
O Genesis Workbench é um Acelerador de Soluções da Databricks em desenvolvimento ativo. Incentivamos as equipes interessadas em usar esta ferramenta a interagir conosco, fornecer feedback e ajudar a moldar nosso roteiro futuro.
(This blog post has been translated using AI-powered tools) Original Post
Receba os posts mais recentes na sua caixa de entrada
Assine nosso blog e receba os posts mais recentes diretamente na sua caixa de entrada.