Simplificación de pipelines de genómica a escala con Databricks Delta
Obtén un avance del nuevo ebook de O'Reilly con la guía paso a paso que necesitas para empezar a usar Delta Lake.
Pruebe este notebook en Databricks
Este blog es el primero de nuestra serie “Genomics Analysis at Scale”. En esta serie, demostraremos cómo la Plataforma de Analítica Unificada para Genómica de Databricks permite a los clientes analizar datos genómicos a escala poblacional. A partir de los resultados de nuestro pipeline de genómica, esta serie proporcionará un tutorial sobre el uso de Databricks para ejecutar el control de calidad de las muestras, la genotipificación conjunta, el control de calidad de cohortes y análisis avanzados de genética estadística.
Desde la finalización del Proyecto Genoma Humano en 2003, ha habido una explosión de datos impulsada por una drástica caída en el costo de la secuenciación de ADN, de 3000 millones de dólares1 para el primer genoma a menos de 1000 dólares en la actualidad.
[1] El Proyecto del Genoma Humano fue un proyecto de $3 mil millones liderado por el Departamento de Energía y los Institutos Nacionales de Salud que comenzó en 1990 y se completó en 2003.
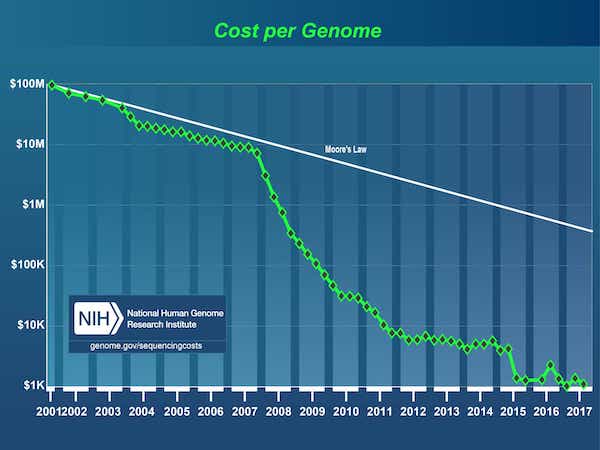
Fuente: Costos de la secuenciación de ADN: Datos
En consecuencia, el campo de la genómica ha madurado hasta un punto en el que las empresas han comenzado a realizar la secuenciación de ADN a escala de población. Sin embargo, la secuenciación del código de ADN es solo el primer paso; los datos en bruto luego deben transformarse a un formato adecuado para el análisis. Generalmente, esto se hace uniendo una serie de herramientas de bioinformática con scripts personalizados y procesando los datos en un solo nodo, una muestra a la vez, hasta obtener una colección de variantes genómicas. Los científicos de bioinformática dedican hoy en día la mayor parte de su tiempo a construir y mantener estos pipelines. A medida que los conjuntos de datos genómicos se han expandido a la escala de petabytes, se ha vuelto un desafío responder de manera oportuna incluso las siguientes preguntas simples:
- ¿Cuántas muestras hemos secuenciado este mes?
- ¿Cuál es el número total de variantes únicas detectadas?
- ¿Cuántas variantes vimos en las diferentes clases de variación?
Para agravar aún más este problema, los datos de miles de personas no se pueden almacenar, rastrear ni versionar y, a la vez, mantenerse accesibles y consultables. En consecuencia, los investigadores suelen duplicar subconjuntos de sus datos genómicos al realizar sus análisis, lo que provoca que el espacio de almacenamiento general y los costos aumenten. En un intento por aliviar este problema, hoy en día los investigadores emplean una estrategia de “congelamiento de datos”, por lo general de seis meses a dos años, en la que detienen el trabajo con datos nuevos y, en su lugar, se centran en una copia congelada de los datos existentes. No existe una solución para desarrollar análisis de forma incremental en períodos de tiempo más cortos, lo que ralentiza el progreso de la investigación.
Existe una necesidad imperiosa de contar con un software robusto que pueda procesar datos genómicos a escala industrial, al tiempo que conserve la flexibilidad necesaria para que los científicos puedan explorar los datos, iterar en sus procesos analíticos y obtener nuevos conocimientos.
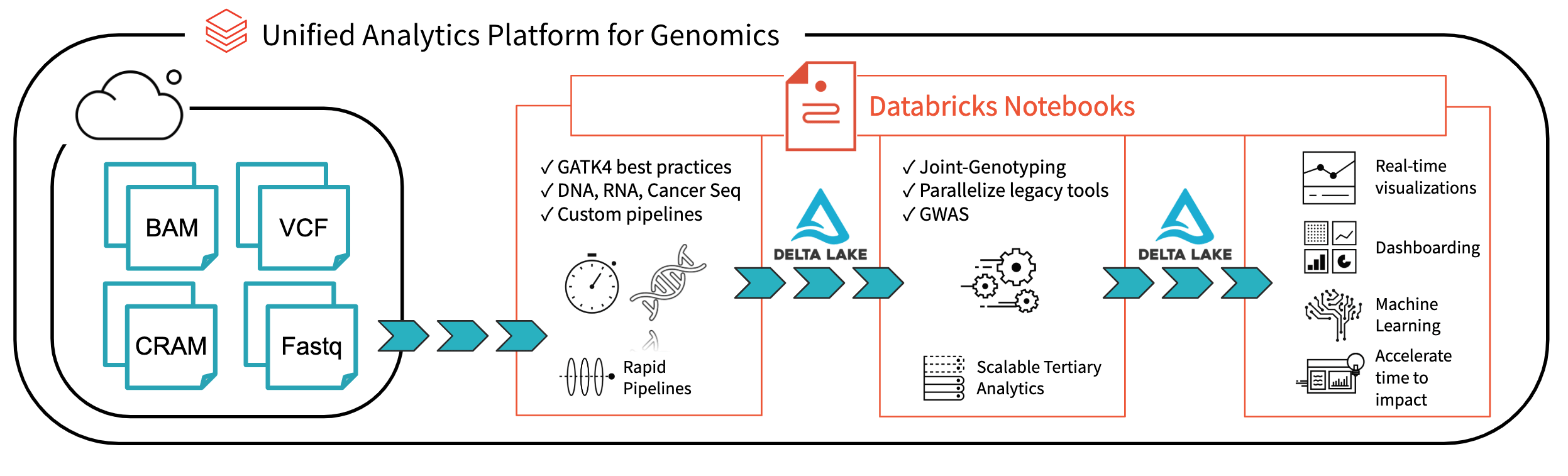
Fig. 1. Arquitectura para el análisis genómico de extremo a extremo con Databricks
Con Databricks Delta: A Unified Management System for Real-time Big Data Analytics, la plataforma Databricks ha dado un paso importante para resolver los problemas de gobernanza, acceso y análisis de datos que enfrentan los investigadores en la actualidad. Con Databricks Delta Lake, puede almacenar todos sus datos genómicos en un solo lugar y crear análisis que se actualicen en tiempo real a medida que se ingieren datos nuevos. En combinación con las optimizaciones de nuestra Unified Analytics Platform for Genomics (UAP4G) para leer, escribir y procesar formatos de archivo de genómica, ofrecemos una solución integral para los flujos de trabajo de los pipelines de genómica. La arquitectura UAP4G ofrece flexibilidad, lo que permite a los clientes integrar sus propias canalizaciones y desarrollar sus propios análisis terciarios. Como ejemplo, hemos destacado el siguiente panel que muestra métricas de control de calidad y visualizaciones que se pueden calcular y presentar de forma automatizada y personalizada para satisfacer sus requisitos específicos.
https://www.youtube.com/watch?v=73fMhDKXykU
En el resto de este blog, repasaremos los pasos que seguimos para crear el panel de control de calidad anterior, que se actualiza en tiempo real a medida que las muestras terminan de procesarse. Al usar un pipeline basado en Delta para procesar datos genómicos, nuestros clientes ahora pueden operar sus pipelines de una manera que proporciona visibilidad en tiempo real, muestra por muestra. Con los notebooks de Databricks (y las integraciones como GitHub y MLflow), pueden hacer un seguimiento y versionar los análisis de una manera que garantizará que sus resultados sean reproducibles. Sus bioinformáticos pueden dedicar menos tiempo al mantenimiento de pipelines y más tiempo a realizar descubrimientos. Vemos el UAP4G como el motor que impulsará la transformación de los análisis ad hoc a la genómica de producción a escala industrial, lo que permitirá una mejor comprensión del vínculo entre la genética y las enfermedades.
Leer datos de muestra
Comencemos por leer los datos de variación de una pequeña cohorte de muestras; la siguiente instrucción lee los datos de un sampleId específico y los guarda usando el formato Delta de Databricks (en la carpeta delta_stream_output).
Ten en cuenta que la carpeta annotations_etl_parquet contiene anotaciones generadas a partir del conjunto de datos de 1000 genomas almacenados en formato parquet. El ETL y el procesamiento de estas anotaciones se realizaron utilizando la plataforma analítica unificada para genómica de Databricks.
Iniciar el streaming de la tabla Delta de Databricks
En la siguiente instrucción, estamos creando el DataFrame de Apache Spark de exomas, que lee una transmisión (a través de readStream) de datos con el formato Delta de Databricks. Este es un DataFrame dinámico o de ejecución continua, es decir, el DataFrame de exomas cargará datos nuevos a medida que se escriban en la carpeta delta_stream_output. Para ver el DataFrame de exomas, podemos ejecutar una consulta de DataFrame para encontrar el recuento de variantes agrupadas por el sampleId.
Al ejecutar la instrucción display, el notebook de Databricks proporciona un panel de streaming para monitorear los trabajos de streaming. Inmediatamente debajo del trabajo de streaming se encuentran los resultados de la instrucción de visualización (es decir, el recuento de variantes por sample_id).
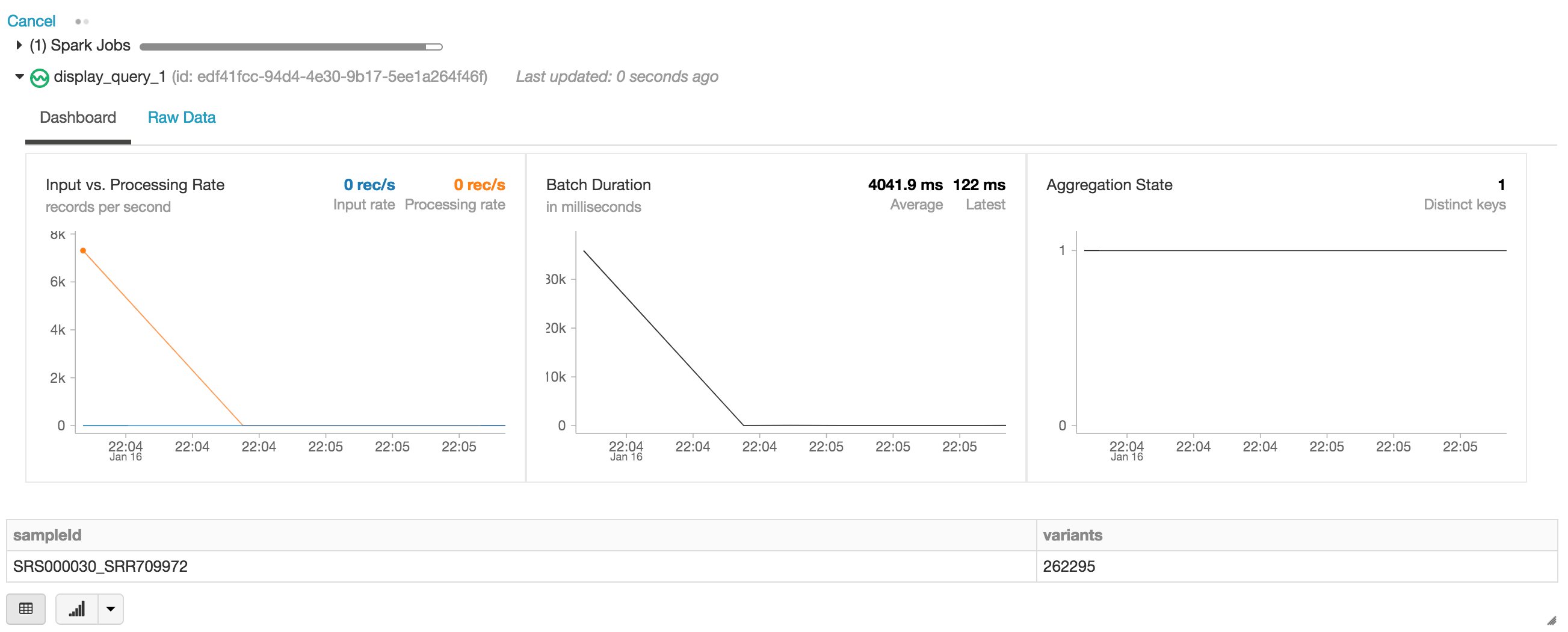
Continuemos respondiendo a nuestro conjunto inicial de preguntas ejecutando otras consultas de DataFrame basadas en nuestro DataFrame exomes.
Recuento de variantes de un solo nucleótido
Para continuar con el ejemplo, podemos calcular rápidamente el número de variantes de un solo nucleótido (SNV), como se muestra en el siguiente gráfico.
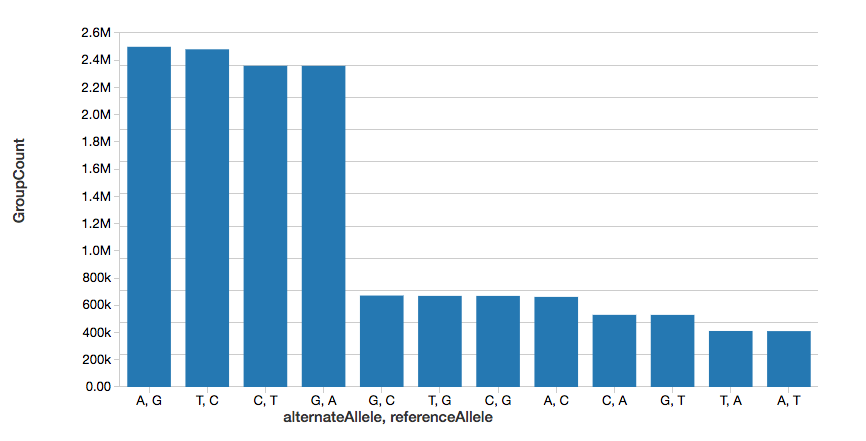
Ten en cuenta que el comando display es parte del espacio de trabajo de Databricks que te permite ver tu DataFrame usando las visualizaciones de Databricks (es decir, sin necesidad de codificar).
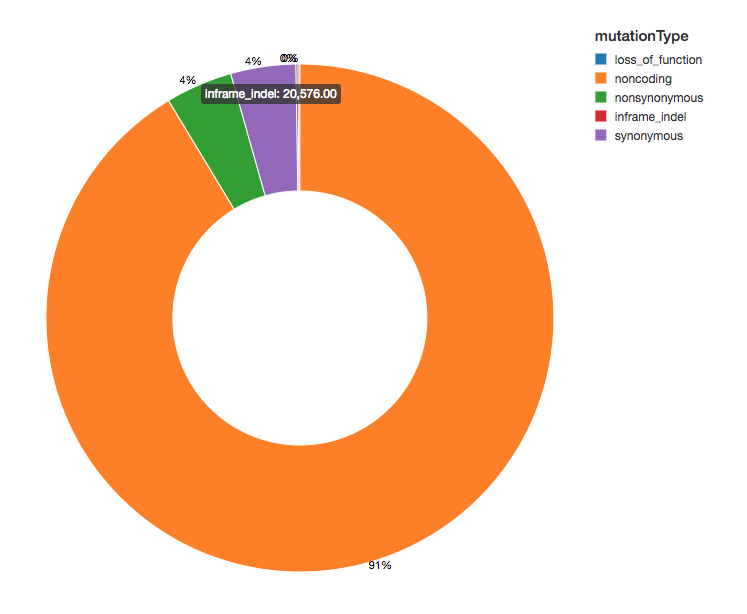
Recuento de variantes
Dado que hemos anotado nuestras variantes con efectos funcionales, podemos continuar nuestro análisis observando la distribución de los efectos de las variantes que vemos. La mayoría de las variantes detectadas flanquean regiones que codifican proteínas; estas se conocen como variantes no codificantes.
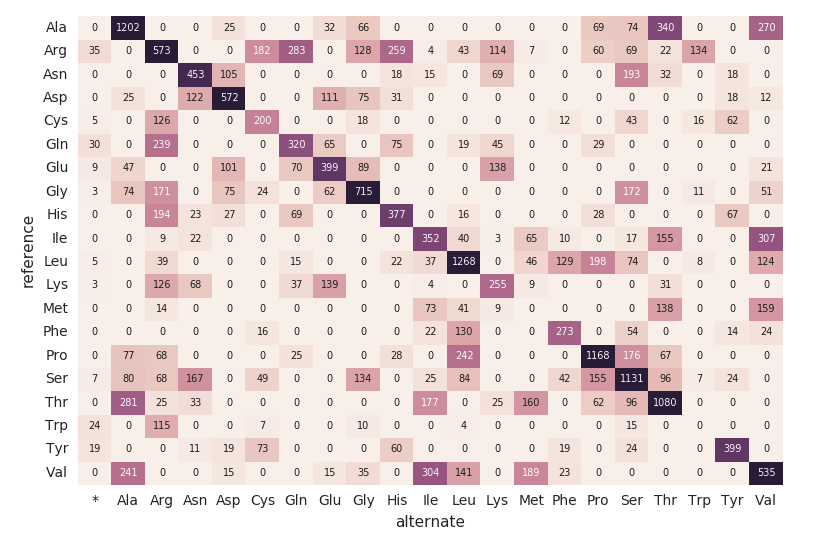
Mapa de calor de sustitución de aminoácidos
Continuando con nuestro DataFrame exomes, calculemos los recuentos de sustitución de aminoácidos con el siguiente fragmento de código. Al igual que los DataFrames anteriores, crearemos otro DataFrame dinámico (aa_counts) para que, a medida que el DataFrame exomes procese nuevos datos, estos se reflejen posteriormente también en los recuentos de sustitución de aminoácidos. También estamos escribiendo los datos en la memoria (es decir, .format(“memory”)) y procesando lotes cada 60 s (es decir, trigger(processingTime=’60 seconds’)) para que el código del mapa de calor de Pandas posterior pueda procesar y visualizar el mapa de calor.
El siguiente fragmento de código lee la tabla de Spark amino_acid_substitutions anterior, determina el recuento máximo, crea una nueva tabla dinámica de Pandas a partir de la tabla de Spark y, luego, grafica el mapa de calor.
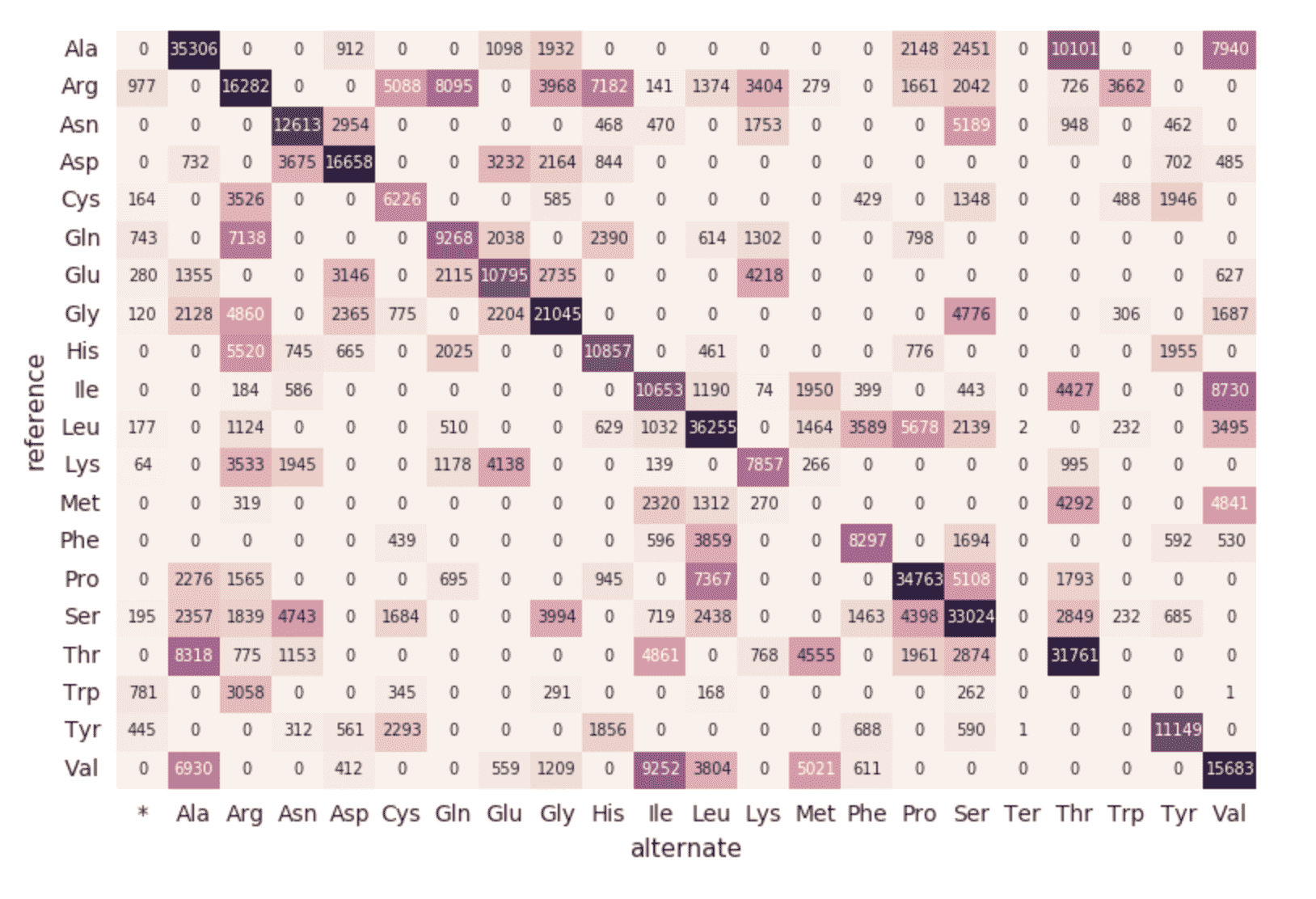
Migración a un proceso continuo
Hasta este momento, los fragmentos de código y las visualizaciones anteriores representan una sola ejecución para un solo sampleId. Pero como estamos usando Structured Streaming y Databricks Delta, este código se puede usar (sin ningún cambio) para construir una canalización de datos de producción que calcule estadísticas de control de calidad de forma continua a medida que las muestras pasan por nuestra canalización. Para demostrar esto, podemos ejecutar el siguiente fragmento de código que cargará todo nuestro conjunto de datos.
Como se describe en los fragmentos de código anteriores, el origen del DataFrame exomes son los archivos cargados en la carpeta delta_stream_output. Inicialmente, habíamos cargado un conjunto de archivos para un solo sampleId (es decir, sampleId = “SRS000030_SRR709972”). El fragmento de código anterior ahora toma todas las muestras parquet generadas (es decir, parquets) y carga de forma incremental esos archivos por sampleId en la misma carpeta delta_stream_output. El siguiente GIF animado muestra la salida abreviada del fragmento de código anterior.
https://www.youtube.com/watch?v=JPngSC5Md-Q
Visualización de su pipeline de genómica
Cuando se desplace de nuevo a la parte superior de su notebook, notará que el DataFrame exomes ahora está cargando automáticamente los nuevos sampleIds. Como el componente de streaming estructurado de nuestro pipeline de genómica se ejecuta continuamente, procesa los datos tan pronto como se cargan nuevos archivos en la carpeta delta_stream_outputpath. Al utilizar el formato Delta de Databricks, podemos garantizar la consistencia transaccional de los datos que se transmiten por streaming al DataFrame de exomas.
https://www.youtube.com/watch?v=Q7KdPsc5mbY
A diferencia de la creación inicial de nuestro DataFrame exomes, observe cómo el panel de supervisión de Structured Streaming ahora está cargando datos (es decir, la “tasa de entrada vs. procesamiento” fluctuante, la “duración del lote” fluctuante y un aumento de claves distintas en el “estado de las agregaciones”). Mientras se procesa el DataFrame exomes, observe las nuevas filas de sampleIds (y los recuentos de variantes). Esta misma acción también se puede observar en la consulta de agrupación por tipo de mutación asociada.
https://www.youtube.com/watch?v=sT179SCknGM
Con Databricks Delta, los nuevos datos son transaccionalmente consistentes en cada paso de nuestro pipeline de genómica. Esto es importante porque garantiza que su canalización sea consistente (mantiene la consistencia de sus datos, es decir, asegura que todos los datos sean “correctos”), confiable (la transacción se completa con éxito o falla por completo) y pueda gestionar actualizaciones en tiempo real (la capacidad de gestionar muchas transacciones simultáneamente, y que ninguna interrupción o falla afecte a los datos). Así, incluso los datos en nuestro mapa de sustitución de aminoácidos descendente (que tuvo una serie de pasos de ETL adicionales) se actualizan sin problemas.
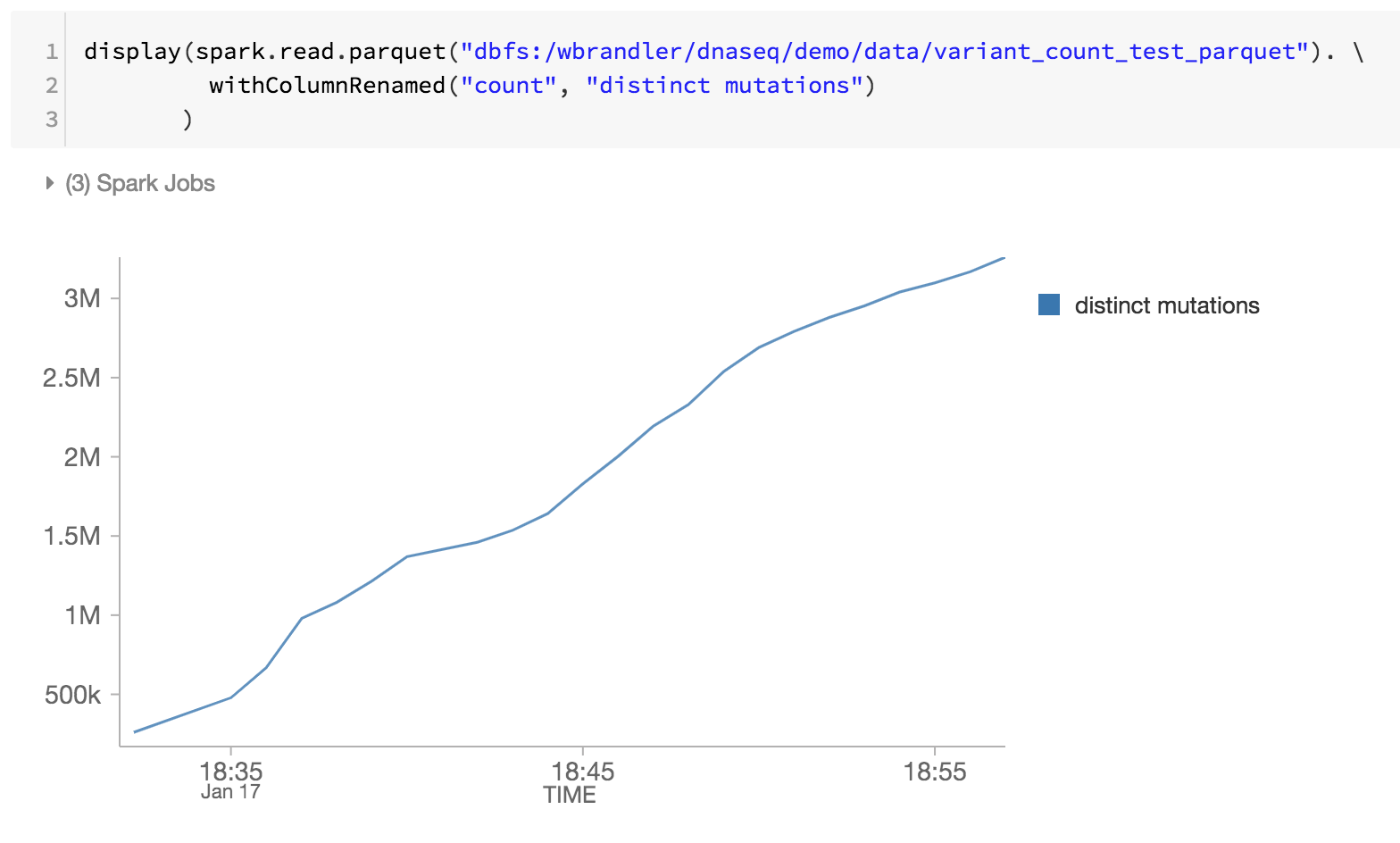
Como último paso de nuestro pipeline genómico, también estamos monitoreando las distintas mutaciones revisando los archivos parquet de Databricks Delta dentro de DBFS (es decir, el aumento de mutaciones distintas a lo largo del tiempo).
Resumen
Usando como base la plataforma de análisis unificado de Databricks, con especial atención en Databricks Delta, los bioinformáticos e investigadores pueden aplicar análisis distribuidos con consistencia transaccional mediante la plataforma de análisis unificado de Databricks para genómica. Estas abstracciones permiten a los profesionales de datos simplificar las canalizaciones de genómica. Aquí hemos creado una canalización de control de calidad de muestras genómicas que procesa datos de forma continua a medida que se procesan nuevas muestras, sin intervención manual. Ya sea que esté realizando ETL o análisis sofisticados, sus datos fluirán a través de su canalización de genómica rápidamente y sin interrupciones. Pruébelo hoy mismo descargando el notebook Simplificación de canalizaciones de genómica a escala con Databricks Delta.
Empiece a analizar la genómica a escala:
- Lea nuestra guía de solucionesde Unified Analytics for Genomics
- Descarga el notebook para simplificar los pipelines genómicos a escala con Databricks Delta
- Regístrese para obtener una prueba gratuita de Databricks Unified Analytics for Genomics
Agradecimientos
Gracias a Yongsheng Huang y a Michael Ortega por sus contribuciones.
Visita el centro en línea de Delta Lake para obtener más información, descargar el código más reciente y unirte a la comunidad de Delta Lake.
Recibe las últimas publicaciones en tu bandeja de entrada
Suscríbete a nuestro blog y recibe las últimas publicaciones directamente en tu bandeja de entrada.