Genesis Workbench: Databricks 기반 생명 과학 애플리케이션을 위한 청사진
작성자: 피터 호킨스, 메이 머클탄, Srijit Chandrashekhar Nair, 일라이 스완슨, 양양, 관위 첸, 람 골리 , 더글라스 무어
제너레이티브 AI는 다양한 생물학적 데이터세트에서 학습된 기본 모델을 활용하여 신약 개발, 예측 모델링, 개인 맞춤형 의학 분야의 고급 애플리케이션을 지원함으로써 생명공학 R&D를 혁신하고 있습니다. 그러나 해당 분야의 과학자들은 종종 상당한 기술 및 관리 장벽에 직면하며, 이로 인해 핵심 연구 활동에 지장을 받을 수 있습니다. 여기에는 GPU 환경 구성, 복잡한 워크플로 관리, 적절한 액세스 제어 보장 등이 포함됩니다. Genesis Workbench는 Databricks Platform에서 생물학용 파운데이션 모델을 사용하여 생명 과학 애플리케이션을 개발하기 위한 청��사진을 제공함으로써 이러한 과제를 해결하기 위한 노력입니다.
AI, 표적 발굴 가속화 및 신약 설계 혁신
생명 과학은 방대한 게놈 및 다중 오믹스 데이터세트와 인공지능(AI) 및 machine learning(ML)의 발전이 융합되면서 근본적인 변환을 경험하고 있습니다. 처음에는 전문적이었던 AI 도구는 단백질 구조 예측, 단백질 언어 모델, 고급 생성 모델의 혁신으로 입증된 정교하고 일반화된 기초 모델로 빠르게 발전했습니다. 방대한 생물학적 서열 및 구조 데이터로 학습된 이러한 기반 모델은 랩 인 더 루프 설계와 같은 프로세스를 통해 신약 개발을 가속화하고 과학자의 일상 업무 효율성을 개선하며 잠재적으로 새로운 설계를 촉진할 수 있도록 지원합니다. 이러한 가속화는 과학 문헌, 전자 건강 기록(EHR), 전자 실험실 노트북(ELN)에서 생물의학 지식을 정확하게 추출하고 합성하는 데 중요한 특수 대규모 언어 모델(LLM)을 통해 더욱 지원됩니다.
안전하고 확장 가능한 연구 플랫폼 구축의 실질적인 과제
표적을 더 잘 찾거나 신약을 설계하는 기술 및 여기에 사용되는 데이터는 매우 엄격하게 보호되는 지적 재산(IP)입니다. 따라서 조직은 데이터와 모델 모두에 대해 개인과 그룹에 적절한 액세스 제어 권한을 부여할 수 있도록 해야 합니다. 모델 사용 감사, 비용 모니터링 등 추가적인 거버넌스 문제는 대규모 생물학적 데이터 세트에 대한 파운데이션 모델의 핵심 고려 사항입니다. 하지만 중요한 점은 이러한 액세스 제어 프로세스가 구현하기 너무 어렵거나 사용자에게 너무 불투명하여 조직 내 과학 커뮤니티의 실질적인 발전을 저해해서는 안 된다는 것입니다. 실제로 많은 조직이 이 균형점을 찾는 데 어려움을 겪습니다.

생물학이나 계산 생물학 분야에 전문 지식이 있음에도 불구하고, 매우 재능 있는 많은 과학자들이 최신 AI 기술의 특정 전문 분야와 관련된 작업 부담 때문에 고급 생물학적 모델을 설정하는 데 어려움을 겪습니다. 이러한 과제에는 대규모 모델을 효율적으로 훈련하는 데 필수적인 GPU 가속을 위한 CUDA 환경 구성과 같은 기술적 복잡성이 포함됩니다. 또한 과학자들은 데이터 처리, 모델 학습 및 MLOps를 자동화하고 효율적으로 확장하는 복잡한 워크플로를 만들고 관리해야 하는 경우가 많습니다. 이러한 작업에는 종종 전통적인 생물학적 훈련 이외의 기술이 필요합니다. 또한 데이터 엔지니어링은 개인정보 보호와 재현성을 유지하기 위해 데이터 거버넌스 정책을 규정 준수하면서 다양한 생물학적 데이터세트를 수집, 정리, 통합해야 하는 중요한 과제를 안고 있습니다. 이러한 비생물학적 요구는 핵심 과학 연구에서 귀중한 시간과 집중력을 빼앗아 생명과학 분야에서 생성 AI 모델을 적용할 때 발전과 혁신을 더디게 합니다. 이러한 격차를 해소하려면 학제 간 협업과 생물학 연구자들의 기술 장벽을 낮추는 도구에 대한 접근성 개선이 필요합니다.
Genesis Workbench: Databricks 기반의 생물학적 AI/ML을 위한 청사진
손쉬운 강력함: Databricks는 데이터와 AI를 쉽게 만듭니다
데이터브릭스는 강력한 거버넌스, 직관적인 사용성, 필요한 모든 데이터 또는 AI 솔루션을 구축할 수 있는 포괄적인 기능을 결합한 통합 분석 플랫폼으로 주목받고 있습니다. 데이터 관리, 보안 및 규정 준수를 위한 중앙 집중식 도구를 통해 모든 기술 수준의 사용자가 쉽게 액세스하면서 데이터를 항상 보호할 수 있습니다. 원활한 협업 기능, 확장 가능한 처리 능력, 모든 데이터, 분석 및 AI 워크로드에 대한 지원으로 엄격한 제어와 단순성을 유지하면서 혁신하고자 하는 조직에 이상적인 기반이 됩니다. 크고 작은 조직 모두 Databricks에서 생물학 모델을 성공적으로 구축했으며, Merck의 TEDDY 기초 모델 제품군부터 Tahoe Therapeutics의 단일 세포 아틀라스까지 다양한 조직이 Databricks에서 생물학 모델을 구축했습니다.
Databricks에서 생명 과학을 강화하는 데 도움이 되는 Genesis Workbench

Genesis Workbench 는 Databricks의 기능을 활용하여 생명 과학 애플리케이션을 개발하기 위한 청사진을 제공합니다. 자동화된 워크플로, GPU 클러스터링, 모델 서빙, MLflow와 같은 기능을 활용하여 AI 기반 생명 과학 연구를 가속화하는 작동 Template을 제공합니다. 사전 패키지된 생물학적 모델과 맞춤형 워크플로를 갖춘 직관적인 Databricks Apps 인터페이스를 특징으로 하여, 과학자들이 복잡한 설정 없이 빠르게 start할 수 있도록 지원합니다.
NVIDIA와 협력하여 디지털 생물학을 위한 생성형 AI 프레임워크인 BioNeMo가 고급 사전 훈련 모델에 쉽게 액세스할 수 있도록 통합되었습니다. BioNeMo 모델은 NVIDIA 하드웨어에 최적화되어 엔터프라이즈 워크로드에 높은 성능과 확장성을 제공합니다.

오픈 소스인 Genesis Workbench는 AI 엔지니어에게 확장 가능한 Template을 제공하여 비생물학적 워크로드를 줄이고, 생물학용 파운데이션 AI 모델을 사용 및 결합하여 신속한 혁신을 촉진합니다. 유니티는 제네시��스 워크벤치를 시작점으로 다양한 모델을 제공하며, 이러한 모델은 앱을 통해 벤치 과학자와 파이프라인을 구축하는 고급 계산 사용자 모두 사용할 수 있습니다. 중요한 것은 APIs에서 모델을 제공함으로써 복잡한 모델 종속성과 GPU 요구 사항을 추상화할 수 있다는 점입니다. 이를 통해 사용자는 일반적으로 사용되지만 매우 복잡한 도구를 단일 파이프라인으로 묶을 수 있습니다.
Genesis workbench는 활발하게 개발 중인 Databricks 솔루션 Accelerator이며, 기능 세트가 계속해서 성장할 것으로 기대합니다.
제네시스 워크벤치 아키텍처
이 애플리케이션은 Databricks 플랫폼을 기반으로 구축되었으며, 다음과 같은 플랫폼 기능을 사용합니다.
- 거버넌스용 Unity Catalog
- UI용 Databricks 앱
- 파운데이션 모델 서빙을 위한 GPU Model Serving
- 확장 가능한 배치 추론 및 미세 조정을 실행하기 위한 레이크플로우 작업
- 대화형 및 배치 워크로드를 위한 GPU가 탑재된 Classic Compute
- 서드파티 라이브러리 통합을 위한 Docker Container Service
- Databricks 자산 Bundle 을 통한 간편한 배포
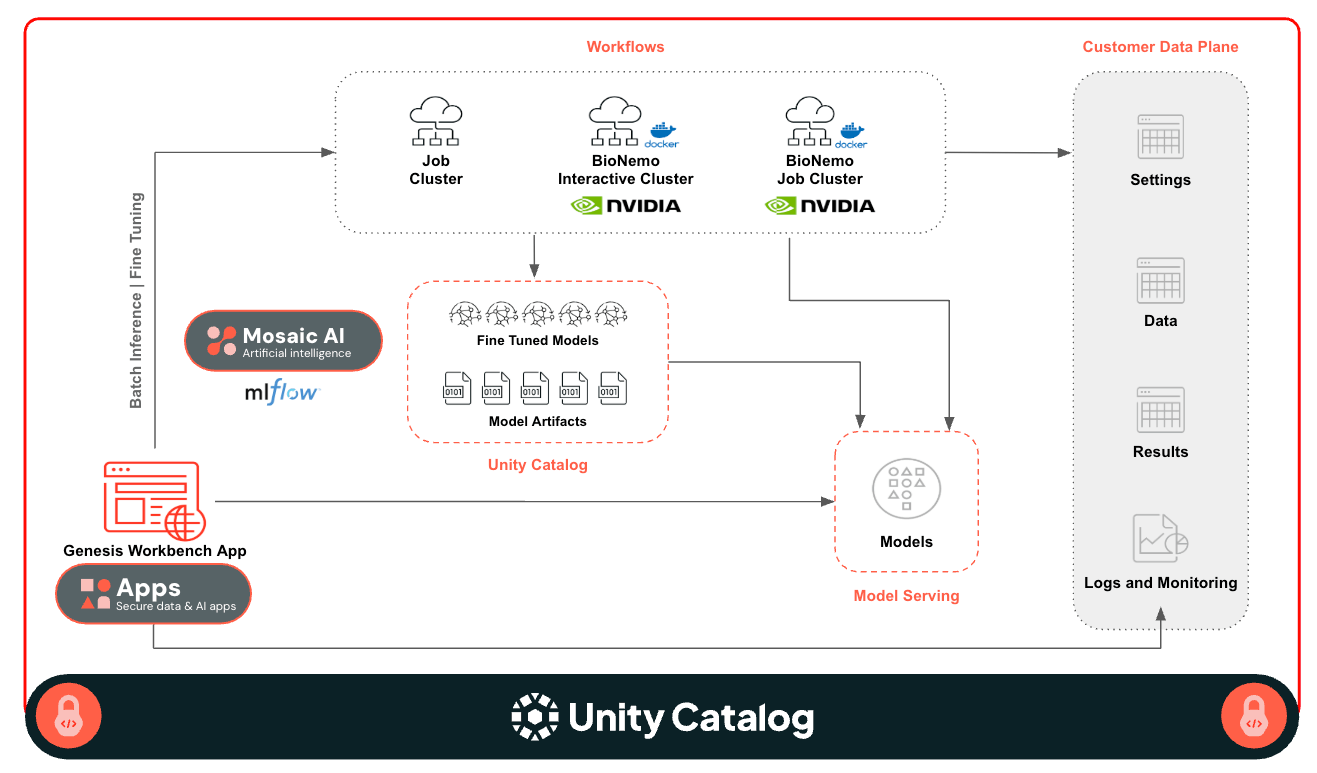
Genesis Workbench의 모듈
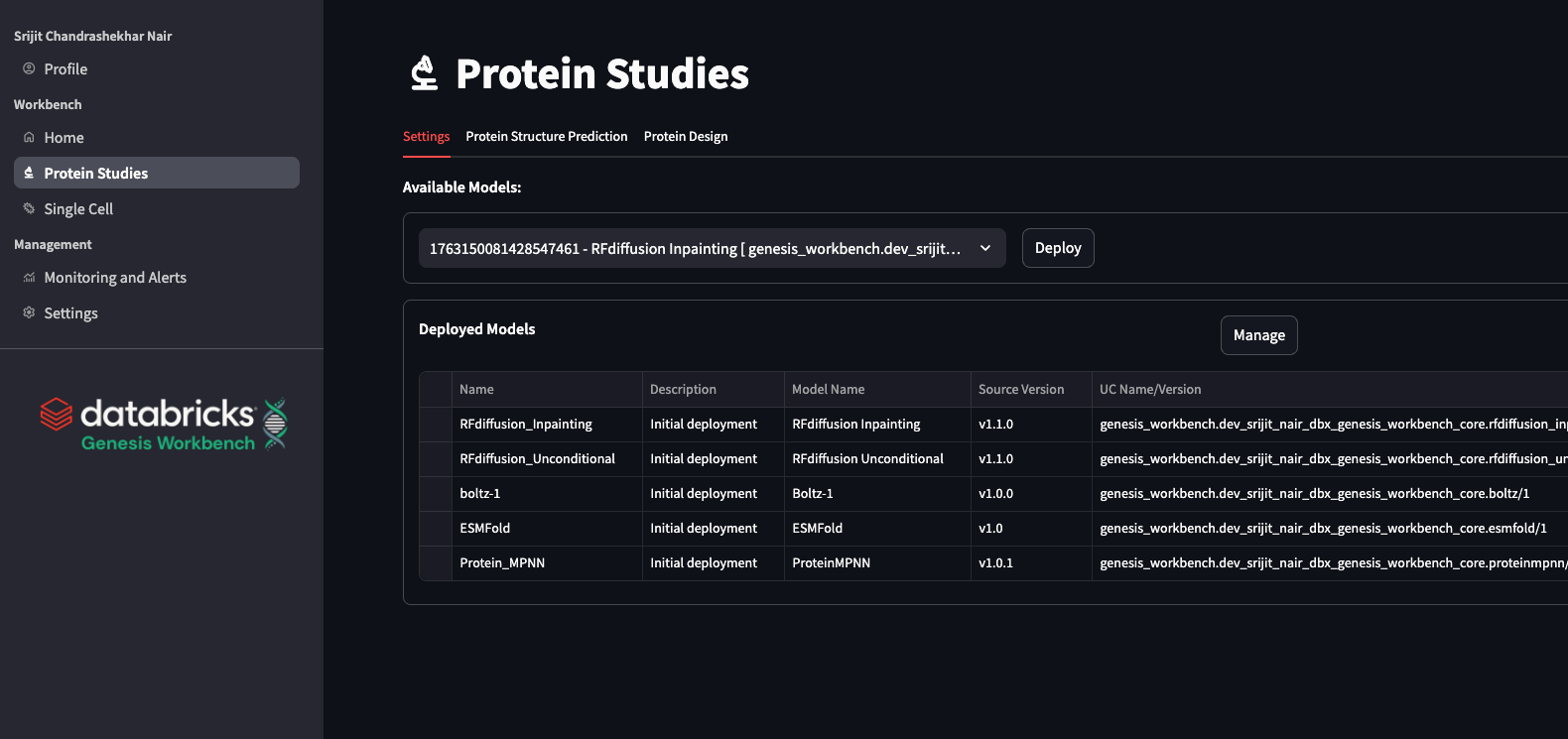
단백질 접기 및 디자인
개요
아미노산 서열로부터 단백질의 3차원 구조를 계산적으로 예측하는 능력은 오랫동안 계산적, 이론적으로 해결해야 할 문제였습니다. CASP 경진대회는 연구 커뮤니티의 모델링 역량을 강화하기 위해 최신 기술을 서로 테스트하는 장이 되어 왔습니다. 딥마인드는 CASP 대회에서 초기 알파폴드 모델(Senior 외, 2020, Nature)과 이후 알파폴드2(Jumper 외, 2021, Nature)를 발표하며 구조 예측 성능을 크게 향상시킬 수 있었습니다. 이를 위해 이전에 측정한 구조로부터 학습하고 대규모 염기서열 데이터베이스의 정보를 통합하는 AI 기술을 활용했습니다. 단백질 멀티머를 포함한 많은 구조를 실험에 가까운 정확도로 예측할 수 있는 능력은 신약 개발 접근 방식에 혁신을 가져왔습니다.
이제 단백질 구조 관련 작업을 위한 모델 동물원이 생겼습니다. 알파폴드-3(Abramsom 외 2024 Nature)(폐쇄형 가중치), 볼츠-1(Wohlend 2024, BioRxiv), 차이-1(차이 발견팀 2024, BioRxiv) 같은 모델은 이제 단백질을 넘어 단백질, DNA, RNA, 저분자 등 보다 진보된 구조로 확장되고 있습니다. Openfold3(Openfold3 팀)와 같은 다른 오픈 모델들이 최근 유사한 기능으로 출시되었으며, 이러한 모델 군은 계속해서 성장할 것으로 예상됩니다. 또한 생성형 모델로 단백질을 생성하는 기능이 빠르게 발전하고 있으며, RFdiffusion(Watson et al. 2023, Nature) 및 ProteinMPNN(Dauparas et al. 2022, Science)과 같은 도구가 이 분야에서 자주 사용됩니다(예: Bielska et al. 2025, front.immunol.), 와 같은 새로운 모델과 BoltzGen(Stark 외. 프리프린트, 레포)과 같은 새로운 모델은 빠르게 진화하는 전산 약물 발견 및 설계를 위한 툴킷을 제공합니다.
Alphafold
알파폴드2의 최신 안정 버전(v2.3.2)이 포함되어 있습니다(Jumper 외, 2021, Nature). 알파폴드 프로세스는 광범위한 기능 계산으로 인해 실행하는 데 시간이 걸리므로, Genesis Workbench에서는 Databricks에서 알파폴드를 워크플로 Job으로 프로비저닝하는 방법을 보여드립니다. 이 job은 CPU가 담당하는 작업(MSA, 특징 추출)과 GPU가 담당하는 폴딩 작업의 두 가지 작업으로 구성됩니다. 이를 통해 각 작업을 적절한 compute 유형에서 실행할 수 있습니다. 이 job은 HPC와 마찬가지로 대기열 시스템을 활용하므로 다른 시퀀스가 대기열에서 대기하는 동안 여러 시퀀스를 동시에 접을 수 있습니다.
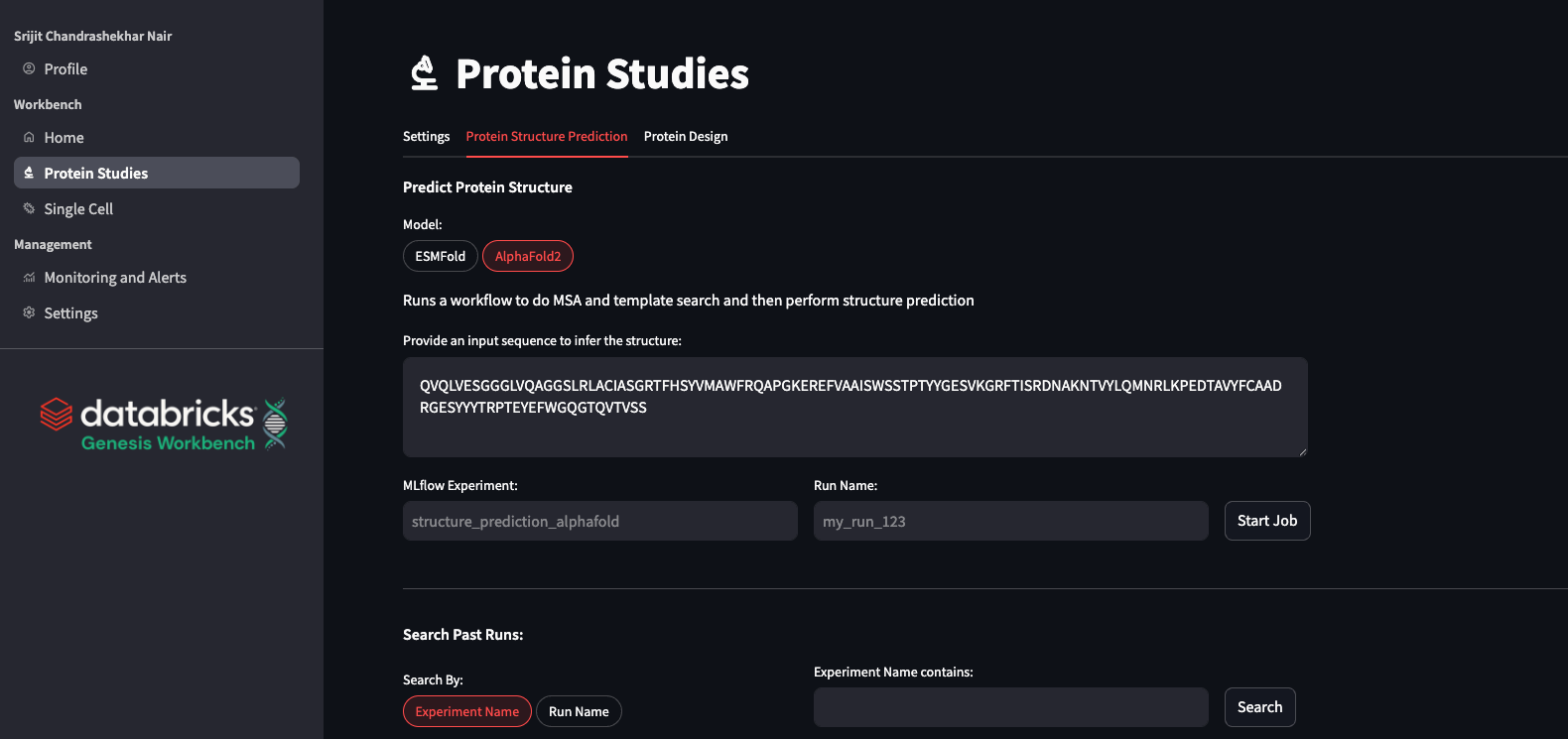
ESMFold
ESMFold(Lin et al. 2023, Science)는 단일 서열에서 단백질 구조를 추론하기 위해 강력한 ESM 단백질 언어 모델과 구조적 헤드를 활용하는 빠르고 비용 효율적인 단백질 구조 예측 딥 러닝 모델입니다. 정확도는 다중 서열 정렬을 사용하는 모델보다 낮지만, ESMFold는 많은 실제 애플��리케이션에 사용하기에 충분합니다. Genesis Workbench에서는 MLflow 모델로 래핑된 ESMfold를 배포하고 Unity Catalog에 등록합니다. Databricks에서는 비용 절감을 위한 scale-to-zero 운영을 지원하는 등 Unity Catalog에 등록된 MLflow 모델을 API로 매우 간단하게 제공할 수 있습니다. ESMFold가 API로 제공되므로 연구원들은 새로운 단백질 구조를 신속하게 예측하고, 이를 계산 파이프라인에 쉽게 통합하며, Genesis Workbench에 표시된 패턴을 사용하여 추가 분석을 위해 결과를 시각화하거나 download할 수 있습니다.
Boltz
Boltz-1(Wohlend 2024, BioRxiv)은 생체 분자 구조 예측을 위한 완전한 오픈 소스 코드 및 가중치(MIT) 모델을 제공합니다. ESMFold와 마찬가지로 Boltz-1을 Unity Catalog에 모델로 등록하고 모델을 제공하는 endpoint에서 서비스합니다. Boltz-1에는 선택 사항인 MSA 입력이 있으며, 이를 생략하거나 미리 계산된 MSA를 사용하거나 mmseqs2(Steinegger, Sölding 2017 Nat.Biotech) 서버 주소를 사용할 수 있습니다. Boltz-1의 다양한 옵션을 보장하기 위해 필수 Boltz-1 모델 구성 요소를 Python 패키지로 래핑하고,alphafold2(Jumper 외, 2021, Nature) 라이브러리의 JackHMMer(Johnson 외, 2010 생물정보학) 옵션을 함께 제공합니다. 이를 통해 사용자는 JackHMMer MSA를 선택할 수 있습니다. default mmseqs2 서버 주소는 공개 URL이라 대부분의 �보안 정책을 위반하므로 이는 일부 사용자에게 유리할 수 있습니다.
Boltz-1 기능 및 MSA 단계는 mlflow 추적 기능으로 래핑됩니다. 추적을 통해 예를 들어 MSA 단계에 대한 추론의 모든 단계와 각 단계의 입력 및 출력을 쉽게 시각화할 수 있습니다. 이 기능은 노트북에서 모델을 사용할 때 특히 유용할 수 있는데, 저장된 파일을 별도로 탐색할 필요 없이 모델에서 어떤 일이 발생했는지 정확히 확인하고 MSA의 문제를 빠르게 파악할 수 있기 때문입니다.
ProteinMPNN 및 RFDiffusion을 사용한 단백질 설계
전산 단백질 설계는 효소 활성과 치료 효과 모두를 위해 효과적인 단백질을 설계하는 방법을 바꿀 수 있는 큰 잠재력을 가진 빠르게 진화하는 분야입니다. RFdiffusion(Watson et al. 2023, Nature) 및 ProteinMPNN(Dauparas et al. 2022, Science)은 예를 들어 계산 항체 설계(Bielska et al. 2025, front.immunol.)와 같은 단백질 설계에 함께 자주 사용됩니다. 그리고 최근에는 신규 항체 설계에도 활용되고 있습니다(Bennett 외, 2025, Nature).
Genesis Workbench에서는 RFDiffusion 및 ProteinMPNN을 제공하는 방법을 시연합니다. 서비스 모델에서 복잡한 종속성을 추상화하면 이러한 모델을 계산 파이프라인으로 쉽게 연결할 수 있는 방법을 보여드립니다. 특히 ESMFold, RFdiffusion, ProteinMPNN을 결합하여 Genesis Workbench 애플리케이션을 호스팅하는 소형 CPU 전용 머신에서 전체 파이프라인을 호출합니다. 더 이상 각 모델의 모든 종속성을 한 곳에 유지 관리하거나 충돌하는 CUDA 버전에 대해 걱정할 필요가 없습니다. 또한 앱에서 출력물을 원래 예측된 구조와 구조적으로 정렬하여 원본 및 설계된 서열을 Mol* 뷰어에 표시합니다.
이는 컴퓨팅 사용자가 Genesis Workbench 내의 구성 요소를 기반으로 조직 또는 연구팀의 목표에 맞게 조정된 프로세스를 생성하기 위해 새로운 툴링을 구축하는 방법을 보여줍니다.
단일 세포 분석
개요
단일 세포 전사체학은 다양한 코호트의 세포 집단을 이해하고, 틈새 세포 유형을 식별하며, 세포 궤적을 밝히는 등 다양한 용도로 활용되는 강력한 기술입니다. 이 분야에서 사용 가능한 데이터의 규모는 샘플 처리의 증가뿐만 아니라 기술의 발전으로 인해 빠르게 증가하고 있습니다. 이는 이러한 규모의 데이터 레이블링 및 처리에 대한 과제를 제시하며 다음과 같은 질문을 던집니다.
- 고급 박사급 연구원이 반복적인 작업에 시간을 소비해야 하는 과도한 부담을 방지하기 위해 분석의 일부를 자동화할 수 있을까요?
- 데이터에 사전 주석을 달아 향후 샘플의 아틀라스 수준 분석을 위한 데이터 탐색을 향상시키려면 어떻게 해야 할까요?
저희는 이러한 과제에 대한 솔루션을 제공하고, 이러한 문제의 변형에 이 접근법들을 적용하기 위한 청사진을 구축�하는 것을 목표로 합니다.
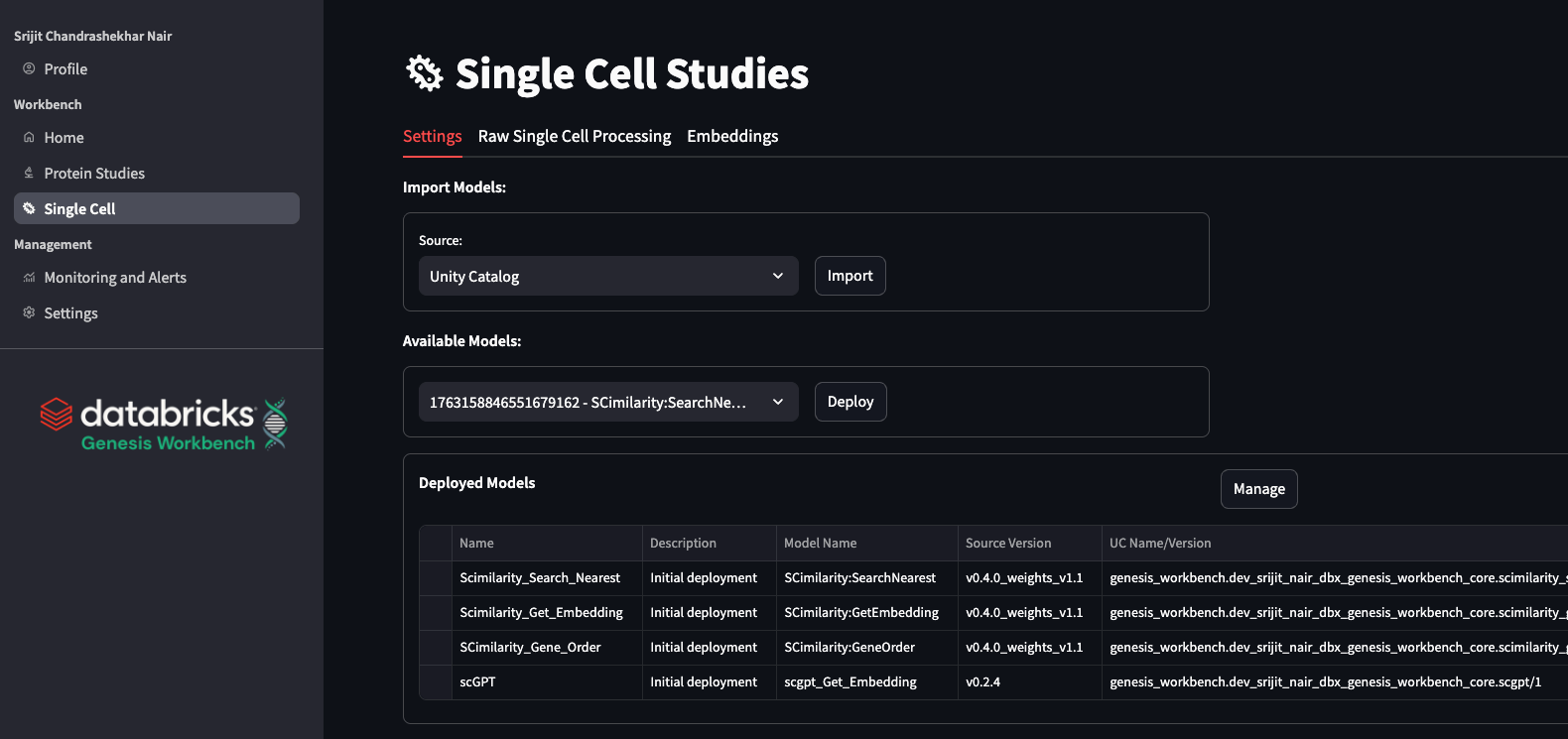
제네시스 워크벤치에서는 단일 셀 처리를 자동화하고 결과를 시각화할 수 있는 툴을 제공하며, MLflow Experiment에서 추적된 모든 결과를 통해 사용자가 데이터를 제어하고 동료와 공유하는 방법을 유지할 수 있습니다. 여기에는 최적의 RAM을 선택하는 CPU 파이프라인과 GPU 가속 워크플로가 포함됩니다. 또한 처리된 데이터에 대한 반응성이 뛰어나고 지연 시간이 짧은 데이터 시각화 기능도 포함되어 있습니다. 또한 단일 세포 발현의 기초 모델도 통합합니다: 아틀라스 수준의 주석, 임베딩 및 검색을 위한 SCimilarity (Heimberg 외, 2024, Nature) 및 scGPT (Cui 외, 2024, Nature Methods)를 통합합니다.
scRNA 데이터 처리: scanpy 및 rapids-singlecell
Genesis Workbench에서는 표준 scanpy (Wolf et al. 2018 Genome Biol) 실행을 위한 워크플로 job을 구성합니다. 이 작업은 최소 RAM 크기를 자동으로 선택하여 사용자 부담과 비용을 동시에 줄입니다. 이 작업은 선택된 parameter뿐만 아니라 다양한 메트릭, 출력, 그림을 단일 MLflow 실행에 Log합니다. 이를 통해 실행 중인 파이프라인에 관한 모든 세부 정보를 자동으로 기록하는 최신 Experiment 추적 절차를 사용할 수 있습니다. 이 Scanpy job 외에도 RAPIDS-singlecell(Dicks et al., 2022, repo, doi) job도 프로비저닝합니다. 이 패키지는 scanpy와 매우 유사하게 작동하고 동일한 parameter 옵션을 갖지만 GPU로 가속화됩니다. 이를 통해 더 빠른 처리량을 확보할 수 있으며, 이는 특정 프로젝트에 필수적입니다.
제네시스 워크벤치 애플리케이션에서는 사용자가 다양한 parameter로 start를 시작하고 애플리케이션 내에서 출력을 확인할 수 있는 간단한 인터페이스를 통해 이러한 파이프라인에 쉽게 액세스할 수 있도록 지원합니다. 지능형 데이터 큐레이션을 수행하여 앱 내에서 결과를 신속하게 분석할 수 있도록 지원하며, 하나의 컴팩트한 애플리케이션 compute에서 여러 사용자를 지원합니다. 그러면 QC 지표, 클러스터, 마커 유전자 발현과 같은 세부 정보를 앱에서 모두 확인할 수 있습니다. 이 프로세싱은 기초 모델과 함께 데이터를 scRNA에 사용하기 전에 중요한 단계가 될 수 있습니다.
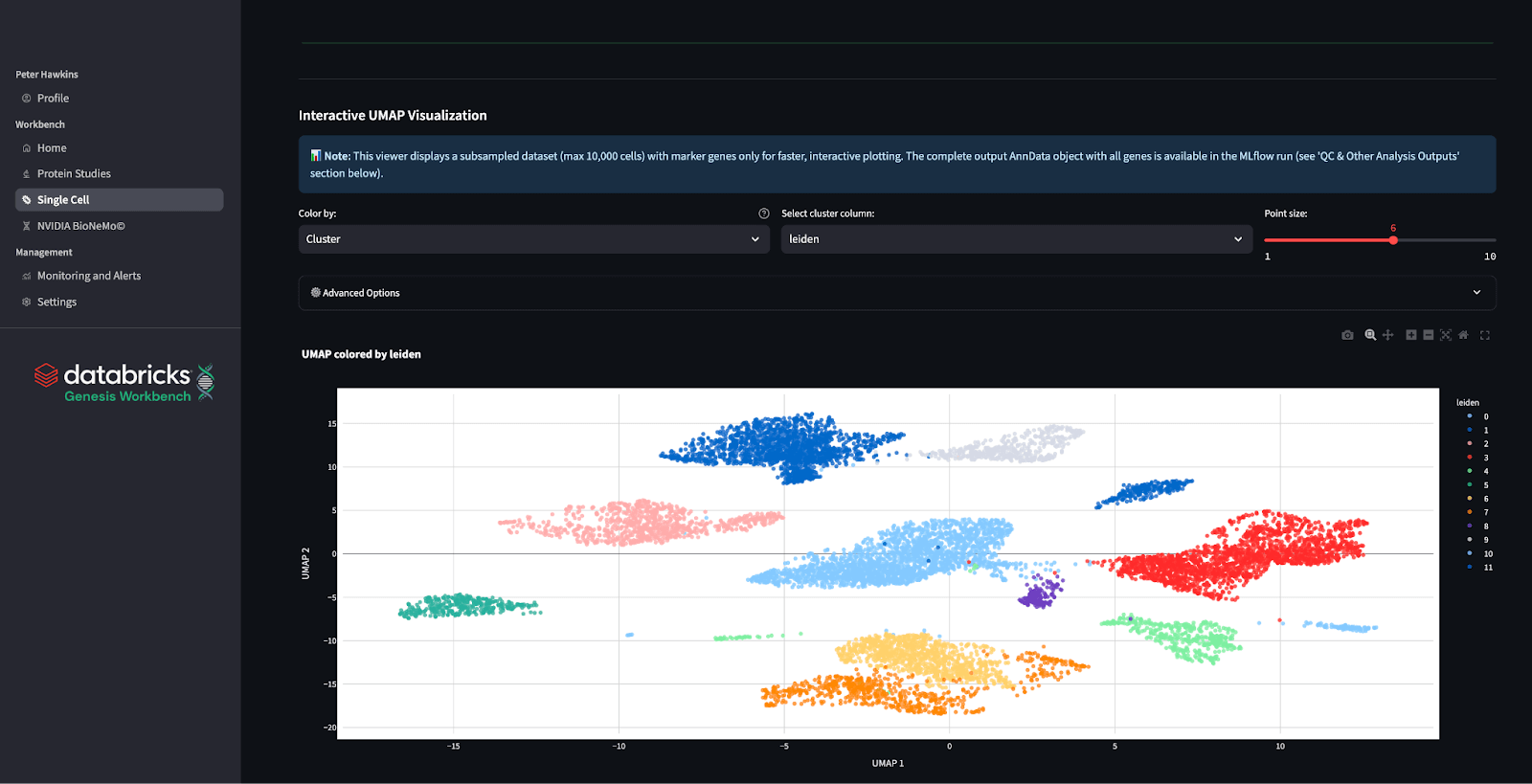
파운데이션 모델: SCimilarity 및 scGPT
Genesis Workbench 내의 단일 세포 분석 모듈은 생명 과학 연구자들이 두 개의 최첨단 파운데이션 모델인SCimilarity 와 scGPT를 이용할 수 있도록 지원합니다.
SCimilarity는 메트릭 학습을 활용하여 대규모 세포 아틀라스를 검색하여 질병 관련 집단과 유사한 체외 모델을 신속하게 식별하고, clusters 없이 참조 카탈로그를 사용하여 장기 및 질병 수준의 정밀도를 달성합니다. 이를 보완하기 위해 scGPT는 섭동 예측, 조절 네트워크 추론, 제로 샷 멀티오믹스 통합을 위해 이중 유전자 세포 임베딩이 포함된 53M parameter 트랜스포머 아키텍처를 사용합니다. 이들은 함께 다양한 데이터세트를 조화시키고, 주석 편향을 최소화하며, 통합 그라디언트를 통한 유사성 및 주의 메커니즘을 통한 scGPT와 같은 기계적인 질병 인사이트를 밝혀내 종양학, 면역학 및 희귀 질환 연구 전반에 걸쳐 중개적 발견과 치료 혁신을 주도합니다(Heimberg 외, 2024, Nature; Cui 외, 2024, Nature Methods).
Genesis Workbench는 사전 구성된 GPU 클러스터, 자동화된 워크플로, MLflow 통합으로 이러한 기본 모델을 패키징하여 두 모델 모두에 대한 주요 Endpoint를 배포합니다: cell_query.gene_order, cell_embedding.get_embeddings, and cell_query.search_nearest SCimilarity용; scGPT용 유전자 임베딩 추론. 예시 노트북은 SCimilarity의 IPF 근섬유아세포 튜토리얼을 기반으로 한 워크플로, 즉 scRNA-seq 데이터 로딩 및 정규화, 임베딩 계산, 제공된 Endpoint를 사용하여 2,340만 개의 셀을 쿼리하여 가장 가까운 질병 농축 집단을 식별하는 과정을 보여줍니다. 식별된 집단은 단일 환경 내에서 유전자 조절 추론, 교란 스크리닝, CRISPR 예측을 위한 다운스트림 scGPT 분석을 가능하게 합니다.
Databricks와 NVIDIA, 속도와 확장성 제공
NVIDIA의 소프트웨어 스택은 가속화된 생명 과학 연구를 위한 강력한 솔루션을 제공합니다. NVIDIA BioNeMo는 바이오 제약을 위한 딥러닝 모델 개발을 가속화하는 오픈 소스 프레임워크입니다. 이를 통해 연구원들은 신속하고 효율적인 도구를 사용하여 DNA, RNA 및 단백질 데이터를 포괄하는 생체 분자 AI 모델을 새로운 차원으로 확장할 수 있습니다. Parabricks는 GPU에 최적화된 알고리즘을 사용하여 빠르고 throughput이 높은 유전체 분석을 제공하므로 유전체학 연구소에서 차세대 시퀀싱 데이터를 신속하게 처리할 수 있습니다. Rapids-SingleCell은 단일 세포 데이터 분석을 향상시켜, GPU 가속을 활용하여 대규모 멀티오믹스 데이�터세트에 대한 확장 가능한 대화형 탐색 및 분석을 가능하게 함으로써 보다 심층적인 생물학적 인사이트를 제공합니다.
엔비디아와 Databricks 엔지니어링 팀 간의 긴밀한 협력을 통해 엔비디아의 고급 소프트웨어 스택을 제네시스 워크벤치에 원활하게 통합하여 생명과학을 위한 신속하고 확장 가능한 AI 기반 워크플로를 제공할 수 있었습니다. 제네시스 워크벤치에서 패턴을 사용하면 가능합니다:
- Databricks Platform 내의 확장 가능한 GPU compute에서 BioNeMo 모델 활용
- BioNeMo 패키지에서 제공하는 모델을 미세 조정하고 대규모 추론에 사용해 보세요.
- 단일 세포 분석을 위한 오픈 소스 GPU 가속 프레임워크인 RAPIDS-singlecell 사용
- 유전체학 분석에 파라브릭 사용
Databricks 대시보드를 사용한 간소화된 가시성 및 비용 최적화
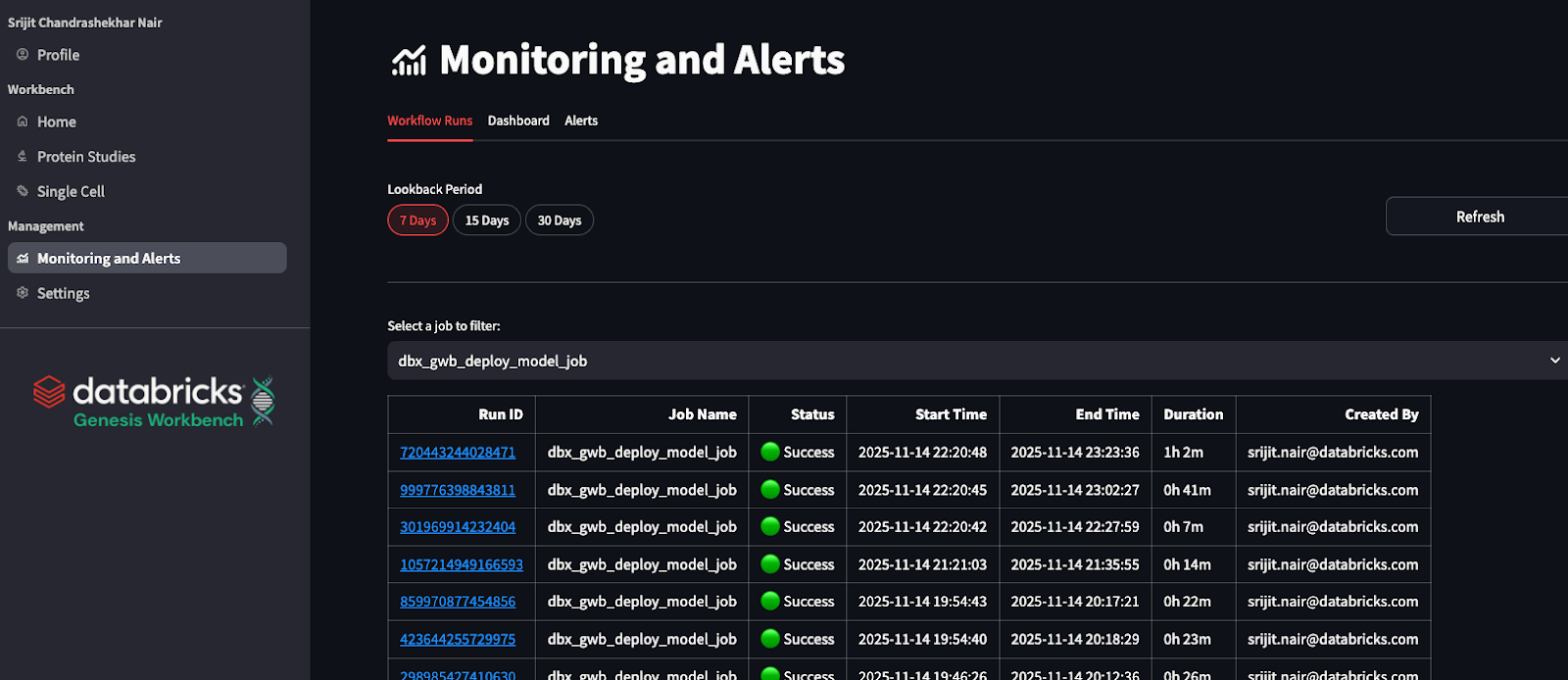
Job 실행 모니터링
장기 실행 백그라운드 프로세스를 필요로 하는 워크플로는 거의 없으며, 이 애플리케이션은 Databricks Workflow를 활용하여 Job을 비동기식으로 시작합니다. Genesis Workbench에는 시스템 내에서 시작된 모든 Databricks 워크플로 Job을 표시하는 전용 모니터링 대시보드가 포함되어 있습니다.
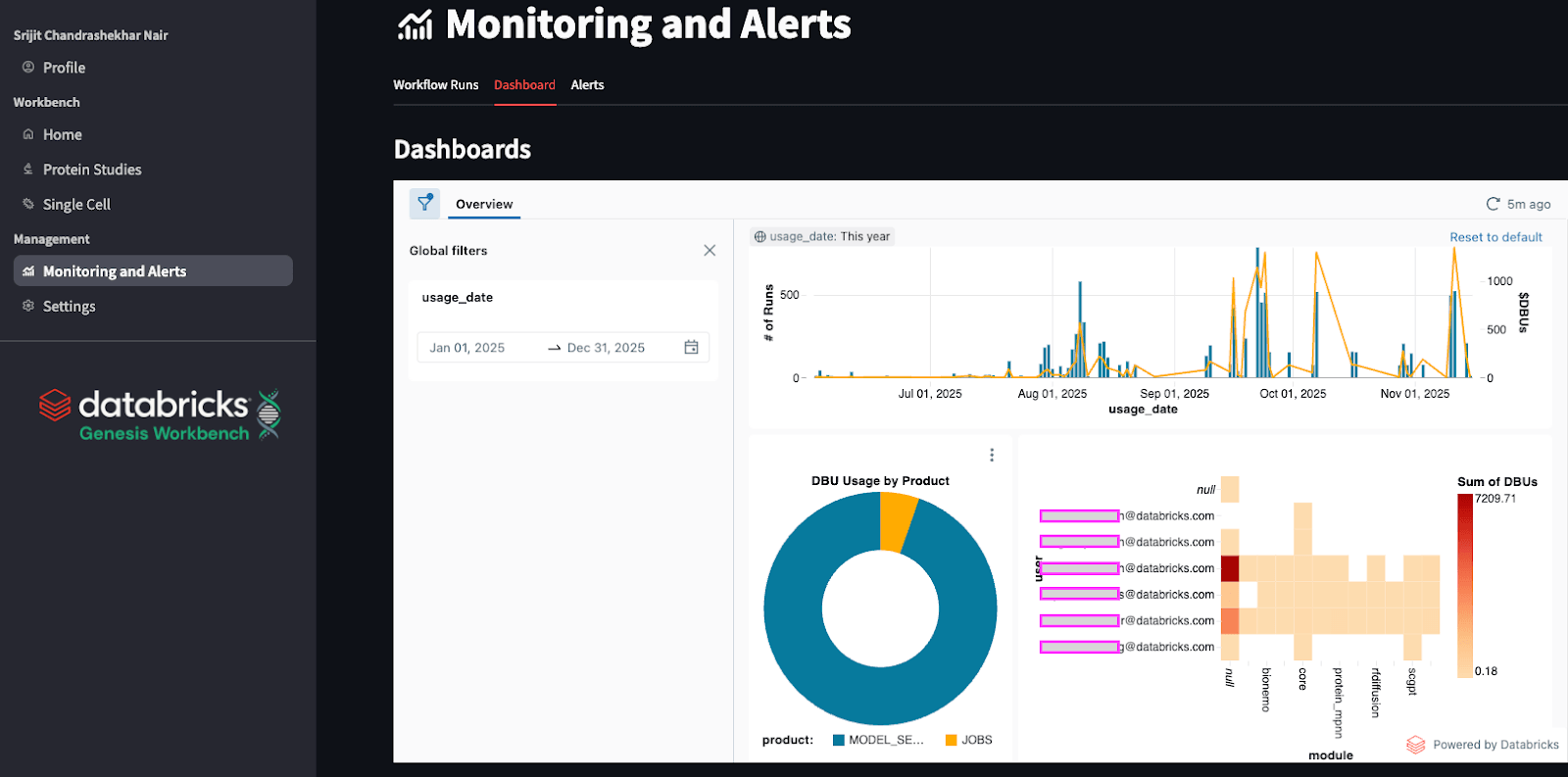
비용 모니터링
기초 모델을 사용하여 계산 파이프라인과 툴을 구축하면 관련 비용에 대한 의문이 제기되는 경우가 많습니다. Databricks는 사용량 및 청구에 대한 정보를 시스템 테이블에 자동으로 캡처합니다. 즉, 이 정보에 대한 대시보드를 쉽게 구축하여 사용량과 지출을 빠르게 보고 분석할 수 있는 사용자 지정 방법을 만들 수 있습니다. 또한 이를 사용하여 과다 사용과 관련된 자동 알림을 생성할 수도 있습니다. 제네시스 워크벤치에서는 애플리케이션 내에서 각 모델에 대한 지출을 시간 및 사용자별로 분류하여 보여주는 대시보드를 자동으로 제공합니다. 또한 대시보드의 형식도 오픈 소스이며, Genesis Workbench를 설치한 팀에서 사용자 지정할 수 있습니다.
다음 단계
설치
제네시스 워크벤치에는 Databricks 자산 번들을 활용하여 애플리케이션을 배포하는 스크립트가 포함되어 있습니다. 제공된 설치 안내 에 따라 GitHub repository 에서 애플리케이션을 download하여 설치할 수 있습니다.
소스에는 다음이 포함됩니다:
- 스크립트를 사용하여 UI 애플리케이션이 포함된 workspace에 Genesis Workbench 코어 모듈을 배포할 수 있습니다.
- 스크립트를 아래 모듈에 배포합니다:
- 단일 세포 모듈
- 단백질 연구 모듈
- 컨테이너 정의 및 �워크플로를 포함하는 BioNeMo 모듈(참고: BioNeMo 컨테이너는 별도로 빌드해야 합니다)
- 모니터링 및 대시보드
피드백
이 프로젝트는 현재 활발히 개발 중이며 여러분의 의견을 기다리겠습니다. 계정 팀에 문의하여 사용 사례를 논의하고 Genesis Workbench에서 설명한 패턴을 어떻게 활용할 수 있는지 알아보세요.
로드맵
- 더 많은 모델 기본 제공
- MSA를 사용한 더 빠른 단백질 폴딩을 위한 고급 MSA 지원
- 단일 세포 데이터에 대한 스트리밍 지원
- BioNemo와 모델 서빙의 통합
- 공간 전사체학 지원
결론
제네시스 워크벤치는 즉시 사용 가능한 MLflow 패키지 생물학적 기초 모델과 Databricks 에코시스템과 원활하게 통합되는 NVIDIA BioNemo 모델을 제공합니다. 이는 Databricks 사용자가 새로운 모델을 Databricks로 가져오고, 여러 모델을 복잡한 파이프라인으로 결합하고, 단일 프레임워크로 벤치 및 계산 과학자를 모두 지원할 수 있는 방법에 대한 청사진입니다.
Unity Catalog를 통한 Databricks의 거버넌스는 모델, Job, 데이터, 애플리케이션에 대한 액세스를 제어하기 위한 일관되고 포괄적인 시스템을 제공합니다. 이를 통해 적절한 액세스 제어를 보장하면서 이러한 파운데이션 모델을 사용하는 애플리케이션 및 데이터 파이프라인의 개발을 획기적으로 간소화할 수 있습니다.
Databricks 모델 서비스 기능을 통해 계산 과학자와 MLOps 엔지니어는 각 모델의 복잡한 종속성과 GPU 요구 사항을 추상화할 수 있습니다. 이를 통해 다른 컴퓨팅 사용자는 종속��성 문제를 신경 쓸 필요 없이 여러 모델을 함께 연결하여 파이프라인을 구축할 수 있습니다. 이를 통해 개발 라이프사이클을 획기적으로 단축할 수 있으며, Unity Catalog 덕분에 적절한 권한 부여를 통해 팀 간에 쉽게 공유할 수 있습니다.
Databricks 앱은 이러한 파운데이션 모델 구성 요소와 생물학적 데이터 뷰어를 기반으로 애플리케이션을 테스트하고 구축하기 위한 빠른 개발자 라이프사이클을 제공합니다. 제네시스 워크벤치 코드는 오픈 소스이므로 각 팀은 팀의 필요에 맞게 앱을 조정하고 개선할 수 있습니다. 필요에 따라 새 모델을 추가하고 Genesis Workbench에서 제공하는 청사진에 따라 비즈니스에 필요한 시스템을 구축할 수 있습니다.
Genesis Workbench 는 활발히 개발 중인 Databricks 솔루션 액셀러레이터입니다. 이 도구 사용에 관심 있는 팀들이 저희와 소통하고, 피드백을 제공하며, 향후 로드맵을 함께 만들어 나가기를 권장합니다.
(이 글은 AI의 도움을 받아 번역되었습니다. 원문이 궁금하시다면 여기를 클릭해 주세요)
최신 게시물을 이메일로 받아보세요
블로그를 구독하고 최신 게시물을 이메일로 받아보세요.