Accélérer la découverte de médicaments : des fichiers FASTA aux informations GenAI sur Databricks
Comment construire un pipeline de bout en bout combinant l'ingénierie des données, les modèles de langage de protéines et la GenAI sur la Databricks Platform.
par Ram Goli et May Merkle-Tan
- Traiter les données biologiques à grande échelle à l'aide des pipelines déclaratifs Lakeflow pour transformer les séquences de protéines brutes au format FASTA en tables prêtes pour l'analyse dans Unity Catalog.
- Classifier les protéines avec des modèles de transformateurs en exploitant ProtBERT, un modèle de langage pour les protéines, afin d'identifier les protéines de transport membranaire, des cibles médicamenteuses clés.
- Query protein insights en langage naturel grâce aux AI Functions qui connectent les LLM directement à vos données, permettant aux chercheurs d'explorer des candidats médicaments prometteurs de manière conversationnelle.
Le développement de médicaments est notoirement lent et coûteux. Le cycle de vie moyen de la Recherche et Développement (R&D) dure de 10 à 15 ans, et une part importante des candidats échoue lors des essais cliniques. L'identification précoce des bonnes cibles protéiques dans le processus a constitué un goulot d'étranglement majeur.
Les protéines sont les "molécules actives" des organismes vivants — elles catalysent les réactions, transportent les molécules et servent de cibles à la plupart des médicaments modernes. La capacité de classifier rapidement les protéines, de comprendre leurs propriétés et d'identifier les candidats sous-étudiés pourrait accélérer considérablement le processus de découverte (par ex. Wozniak et al., 2024, Nature Chemical Biology).
C'est là que la convergence de l'ingénierie des données, du machine learning (ML) et de l'IA générative devient transformative. En fait, vous pouvez construire l'intégralité de ce pipeline sur une seule plateforme : la Databricks Data Intelligence Platform.
Ce que nous construisons
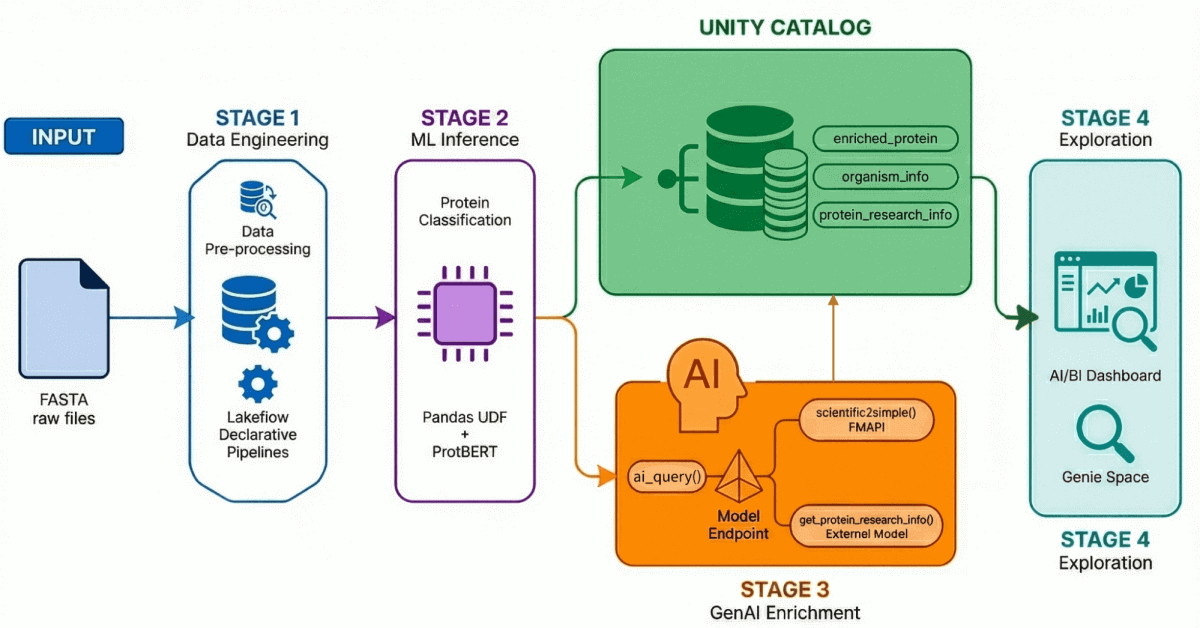
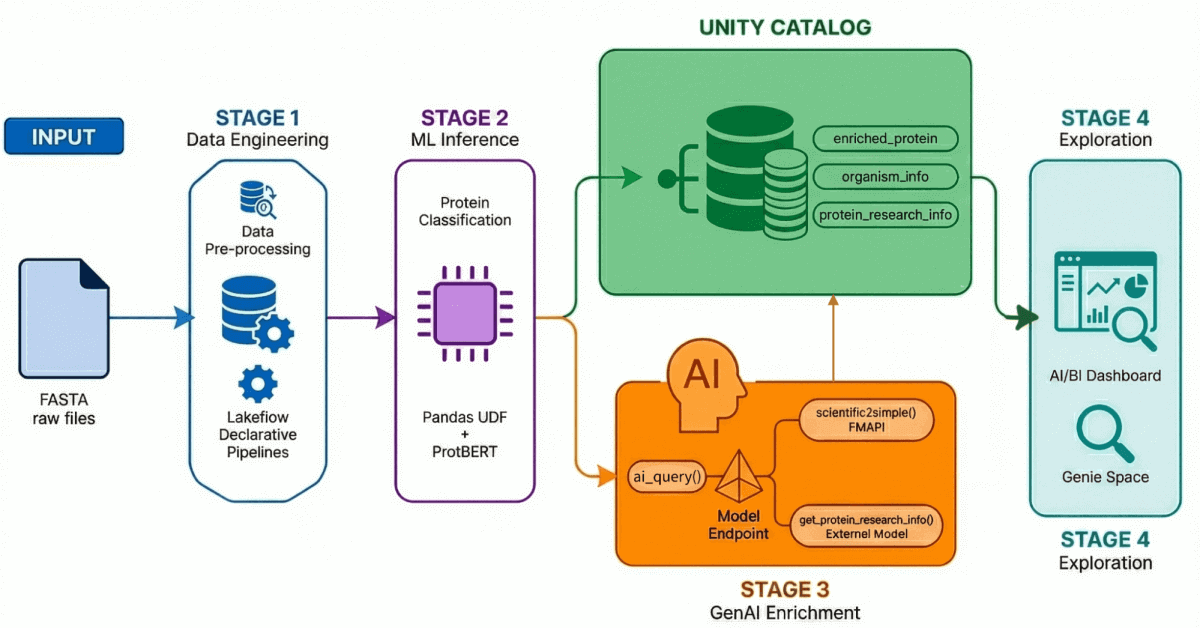
Notre accélérateur de solution pour la découverte de médicaments basée sur l'IA démontre un workflow de bout en bout à travers quatre processus clés :
- Ingestion et traitement des données : plus de 500 000 séquences de protéines sont ingérées et traitées à partir d'UniProt.
- Classification optimisée par l'IA : Un modèle transformer est utilisé pour classifier ces protéines comme étant soit hydrosolubles, soit de transport membranaire.
- Génération d'insights : les données sur les protéines sont enrichies d'insights de recherche générés par les LLM.
- Exploration en langage naturel : toutes les données traitées et enrichies sont accessibles via un tableau de bord et un environnement basés sur l'IA qui prennent en charge les requêtes en langage naturel.
Parcourons chaque étape :
{kind=link}
Étape 1 : Ingénierie des données avec les pipelines déclaratifs Lakeflow
Les données biologiques brutes arrivent rarement dans un format propre et prêt pour l'analyse. Nos données source se présentent sous la forme de fichiers FASTA, un format standard pour la représentation des séquences protéiques, qui ressemble à ceci :
Pour un œil non averti, ces données de séquence sont presque impossibles à interpréter : une chaîne dense de codes d'acides aminés à une seule lettre. Pourtant, à la fin de ce pipeline, les chercheurs peuvent interroger ces mêmes données en langage naturel, en posant des questions comme « Affichez les protéines membranaires humaines peu étudiées avec un indice de confiance de classification élevé » et recevoir en retour des informations exploitables.
À l'aide des pipelines déclaratifs Lakeflow, nous construisons une architecture médaillon qui affine progressivement ces données :
- Couche Bronze : Ingestion brute de fichiers FASTA à l'aide de BioPython, en extrayant les ID et les séquences.
- Couche Silver : Analyse et structuration — nous extrayons les noms de protéines, les informations sur les organismes, les noms de gènes et d'autres métadonnées à l'aide de transformations regex.
- Couche Gold/Enrichie : données organisées prêtes pour l'analyse, enrichies de métriques dérivées telles que le poids moléculaire, et prêtes pour les tableaux de bord, les modèles de ML et la recherche en aval. Il s'agit de la couche de confiance que les analystes et les scientifiques interrogent directement.
Le résultat : Des données de protéines propres et gouvernées dans Unity Catalog, prêtes pour le ML et l'analytique en aval. Fait crucial, le data lineage qui s'étend au-delà de cette étape jusqu'aux autres étapes (mises en évidence ci-dessous) apporte une valeur incroyable à la reproductibilité scientifique.
Étape 2 : Classification des protéines avec les modèles Transformer
Toutes les protéines ne se valent pas en matière de découverte de médicaments. Les protéines de transport membranaire — celles qui sont intégrées dans les membranes cellulaires — sont des cibles médicamenteuses particulièrement importantes car elles contrôlent ce qui entre dans les cellules et ce qui en sort.
Nous exploitons ProtBERT-BFD, un modèle de langage de protéines basé sur BERT du Rostlab, spécifiquement affiné pour la classification des protéines membranaires. Ce modèle traite les séquences d'acides aminés comme un langage et apprend les relations contextuelles entre les résidus pour prédire la fonction des protéines.
Le modèle génère une classification (membranaire ou soluble) ainsi qu'un score de confiance, que nous réécrivons dans Unity Catalog pour le filtrage et l'analyse en aval.
Étape 3 : Enrichir des données avec la GenAI
La classification nous indique ce qu'est une protéine. Mais les chercheurs doivent savoir pourquoi c'est important : quelles sont les recherches récentes ? Où sont les lacunes ? S'agit-il d'une cible médicamenteuse sous-explorée ?
C'est là que les LLM entrent en jeu. En exploitant à la fois l'API Foundational Model de Databricks et les points de terminaison de modèles externes, nous créons des AI Functions enregistrées qui enrichissent les enregistrements de protéines avec un contexte de recherche.
Étape 4 : exploration en langage naturel
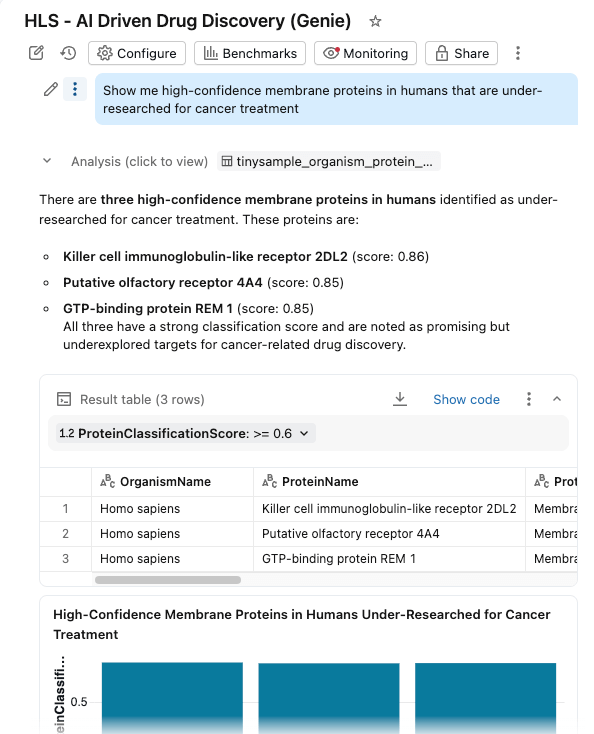
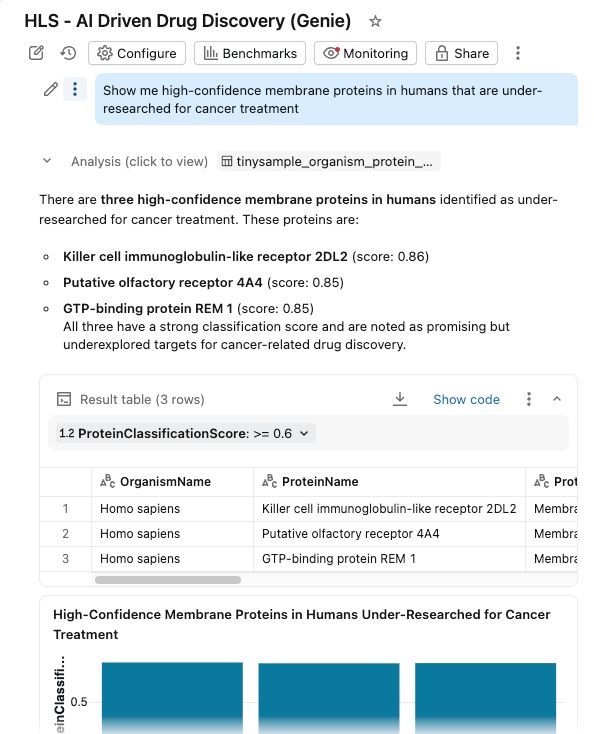
Nous regroupons le tout dans un tableau de bord AI/BI avec Genie Space activé.
Les chercheurs peuvent désormais :
- Filtrer les protéines par organisme, score de classification et type de protéine
- Explorez les distributions des poids moléculaires et de la confiance de la classification.
- Posez des questions en langage naturel : "Montrez-moi les protéines membranaires de haute confiance chez l'homme qui sont sous-étudiées pour le traitement du cancer"
{kind=link}
Le tableau de bord interroge les mêmes tables gouvernées dans Unity Catalog, les Fonctions d'IA fournissant un enrichissement à la demande (ou par lots).
La puissance d'une plateforme unifiée
Ce qui rend cette solution si convaincante ne tient pas à un seul de ses composants, mais au fait que tout s'exécute sur une seule et même plateforme :
| Compétence | Fonctionnalité Databricks |
|---|---|
| Ingestion de données et ETL | Pipelines déclaratifs Lakeflow |
| Gouvernance des données | Unity Catalog |
| Inférence ML | GPU compute |
| Intégration LLM | FMAPI + Modèles externes + Fonctions d'IA |
| Analytique | Databricks SQL |
| Exploration | Tableaux de bord IA/BI + Genie Space |
Point essentiel : aucun mouvement de données n'est effectué entre les systèmes. Aucune infrastructure MLOps distincte. Pas d'outils de BI déconnectés. La séquence protéique qui entre dans le pipeline passe par la transformation, la classification, l'enrichissement et finit par être interrogeable en langage naturel, le tout dans le même environnement géré.
L'accélérateur de solution complet est disponible sur GitHub :
github.com/databricks-industry-solutions/ai-driven-drug-discovery
Et après ?
Cet accélérateur démontre l'art du possible. En production, vous pourriez l'étendre à :
- Traiter la base de données UniProt complète avec des Endpoints à throughput provisionné
- Ajouter d'autres modèles de classification (open source ou personnalisés) pour les différentes propriétés des protéines
- Créer des pipelines RAG sur la base de la littérature scientifique pour obtenir des réponses de LLM plus fondées
- Intégration avec les workflows de simulation moléculaire en aval
- Se connecter à la prédiction de la structure des protéines (AlphaFold/ESMFold) pour ajouter un contexte structurel 3D aux protéines classifiées
- Extension à d'autres formats génomiques (FASTQ, VCF, BAM) à l'aide de Glow pour le séquençage et l'analyse de variants à grande échelle
Les fondations sont en place. La plateforme est unifiée. La seule limite est la science que vous souhaitez accélérer. Lancez-vous dès aujourd'hui !
(Cet article de blog a été traduit à l'aide d'outils basés sur l'intelligence artificielle) Article original
Recevez les derniers articles dans votre boîte mail
Abonnez-vous à notre blog et recevez les derniers articles directement dans votre boîte mail.