Beschleunigung der Wirkstoffentdeckung: Von FASTA-Dateien zu GenAI-Erkenntnissen auf Databricks
Wie man eine End-to-End-Pipeline erstellt, die Data Engineering, Protein-Sprachmodelle und GenAI auf der Databricks Platform kombiniert.
von Ram Goli und May Merkle-Tan
- Biologische Daten auf Scale mit Lakeflow Declarative Pipelines verarbeiten, um rohe FASTA-Proteinsequenzen in analysebereite Tabellen im Unity Catalog umzuwandeln.
- Proteine mit Transformer-Modellen klassifizieren, indem ProtBERT, ein Protein-Sprachmodell, genutzt wird, um Membrantransportproteine – wichtige Wirkstoffziele – zu identifizieren.
- Fragen Sie Proteinerkenntnisse in natürlicher Sprache über AI Functions ab, die LLMs direkt mit Ihren Daten verbinden, sodass Forschende vielversprechende Wirkstoffkandidaten in einem dialogorientierten Format untersuchen können.
Die Arzneimittelentwicklung ist bekanntermaßen langsam und teuer. Der durchschnittliche Lebenszyklus von Forschung und Entwicklung (F&E) erstreckt sich über 10–15 Jahre, wobei ein erheblicher Teil der Kandidaten in klinischen Studien scheitert. Ein wesentlicher Engpass bestand darin, die richtigen Zielproteine frühzeitig im Prozess zu identifizieren.
Proteine sind die "Arbeitsmoleküle" lebender Organismen – sie katalysieren Reaktionen, transportieren Moleküle und dienen als Zielmoleküle für die meisten modernen Medikamente. Die Fähigkeit, Proteine schnell zu klassifizieren, ihre Eigenschaften zu verstehen und wenig erforschte Kandidaten zu identifizieren, könnte den Entdeckungsprozess drastisch beschleunigen (z. B. Wozniak et al., 2024, Nature Chemical Biology).
Genau hier wird die Konvergenz von Data Engineering, maschinellem Lernen (ML) und generativer KI transformativ. Tatsächlich können Sie diese gesamte Pipeline auf einer einzigen Plattform erstellen – der Databricks Data Intelligence Platform.
Woran wir arbeiten
Unser KI-Driven Drug Discovery Solution Accelerator demonstriert einen End-to-End-Workflow anhand von vier Schlüsselprozessen:
- Datenaufnahme und -verarbeitung: Über 500.000 Proteinsequenzen werden von UniProt aufgenommen und verarbeitet.
- KI-gestützte Klassifizierung: Ein Transformer-Modell wird verwendet, um diese Proteine entweder als wasserlöslich oder als Membrantransport zu klassifizieren.
- Erkenntnisgewinnung: Proteindaten werden mit LLM-generierten Forschungserkenntnissen angereichert.
- Exploration in natürlicher Sprache: Alle verarbeiteten und angereicherten Daten werden über ein KI-gestütztes Dashboard und eine Umgebung zugänglich gemacht, die Abfragen in natürlicher Sprache unterstützt.
Gehen wir die einzelnen Phasen durch:
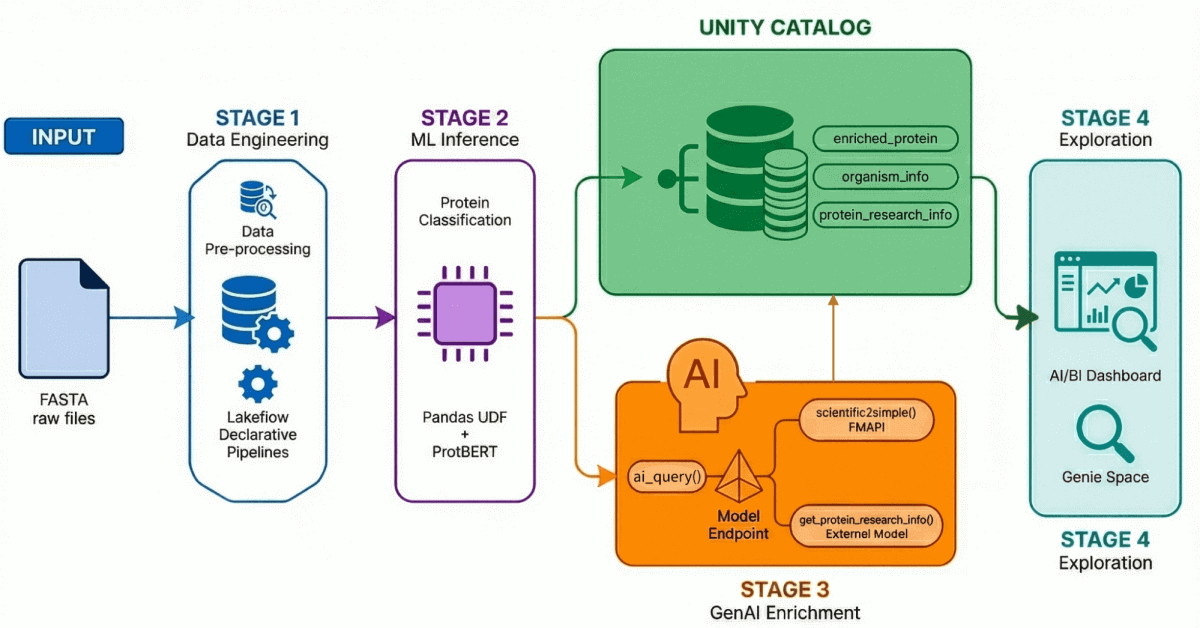
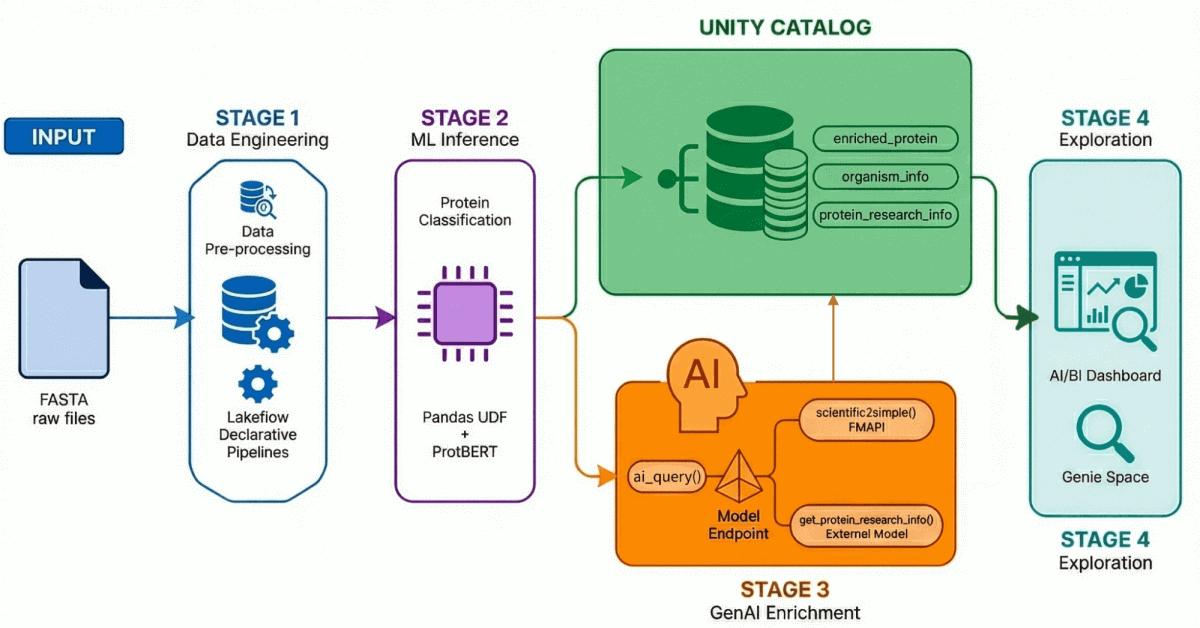{kind=link}
Stufe 1: Data Engineering mit deklarativen Lakeflow-Pipelines
Biologische Rohdaten liegen selten in einem sauberen, analysebereiten Format vor. Unsere Quelldaten liegen als FASTA -Dateien vor – ein Standardformat zur Darstellung von Proteinsequenzen, das etwa so aussieht:
Für das ungeschulte Auge sind diese Sequenzdaten nahezu unmöglich zu interpretieren – eine dichte Zeichenfolge aus Einbuchstaben-Aminosäurecodes. Doch am Ende dieser Pipeline können Forschende dieselben Daten in natürlicher Sprache abfragen, indem sie Fragen stellen wie „Zeig mir wenig erforschte Membranproteine beim Menschen mit hoher Klassifizierungssicherheit“, und erhalten im Gegenzug handlungsorientierte Erkenntnisse.
Mithilfe von deklarativen Lakeflow-Pipelines erstellen wir eine Medallion-Architektur, die diese Daten schrittweise verfeinert:
- Bronze-Schicht: Rohe Erfassung von FASTA-Dateien mit BioPython, wobei IDs und Sequenzen extrahiert werden.
- Silber-Schicht: Parsen und Strukturieren – wir extrahieren Proteinnamen, Organismusinformationen, Gennamen und andere Metadaten mithilfe von Regex-Transformationen.
- Gold/Angereicherte Schicht: Kuratierte, analysebereite Daten, die mit abgeleiteten Metriken wie dem Molekulargewicht angereichert sind – bereit für Dashboards, ML-Modelle und nachgelagerte Forschung. Dies ist der Trusted Layer, den Analysten und Data Scientists direkt abfragen.
Das Ergebnis: Saubere, verwaltete Proteindaten im Unity Catalog, bereit für nachgelagertes ML und Analysen. Entscheidend ist, dass die Datenherkunft, die sich über diese Phase hinaus auf die anderen Phasen (unten hervorgehoben) erstreckt, einen unglaublichen Wert für die wissenschaftliche Reproduzierbarkeit bietet.
Stufe 2: Proteinklassifizierung mit Transformer-Modellen
Bei der Wirkstoffentdeckung sind nicht alle Proteine gleichwertig. Membrantransportproteine – also solche, die in Zellmembranen eingebettet sind – sind besonders wichtige Zielstrukturen für Medikamente, da sie steuern, was in die Zellen ein- und austritt.
Wir nutzen ProtBERT-BFD, ein BERT-basiertes Protein-Sprachmodell vom Rostlab, das speziell für die Klassifizierung von Membranproteinen feinabgestimmt wurde. Dieses Modell behandelt Aminosäuresequenzen wie Sprache und lernt kontextuelle Beziehungen zwischen Resten, um die Proteinfunktion vorherzusagen.
Das Modell gibt eine Klassifizierung (als Membran oder löslich) zusammen mit einem Konfidenzwert aus, die wir zur nachgelagerten Filterung und Analyse in den Unity Catalog zurückschreiben.
Stufe 3: Daten anreichern mit GenAI
Die Klassifizierung sagt uns, was ein Protein ist. Aber Forscher müssen wissen, warum es wichtig ist– was sind die neuesten Forschungsergebnisse? Wo gibt es Lücken? Ist dies ein wenig erforschtes Wirkstoffziel?
Hier kommen LLMs ins Spiel. Durch die Nutzung der Foundational Model API von Databricks sowie der Endpunkte für externe Modelle erstellen wir registrierte AI Functions, die Proteindatensätze mit Forschungskontext anreichern.
Stufe 4: Exploration in natürlicher Sprache
Wir führen alles in einem KI/BI-Dashboard mit aktiviertem Genie Space zusammen.
Forschende können jetzt:
- Proteine filtern nach Organismus, Klassifizierungsscore und Proteintyp
- Verteilungen untersuchen von Molekulargewichten und Klassifizierungskonfidenz
- Stellen Sie Fragen in natürlicher Sprache: "Zeigen Sie mir Membranproteine mit hoher Konfidenz beim Menschen, die für die Krebsbehandlung wenig erforscht sind"
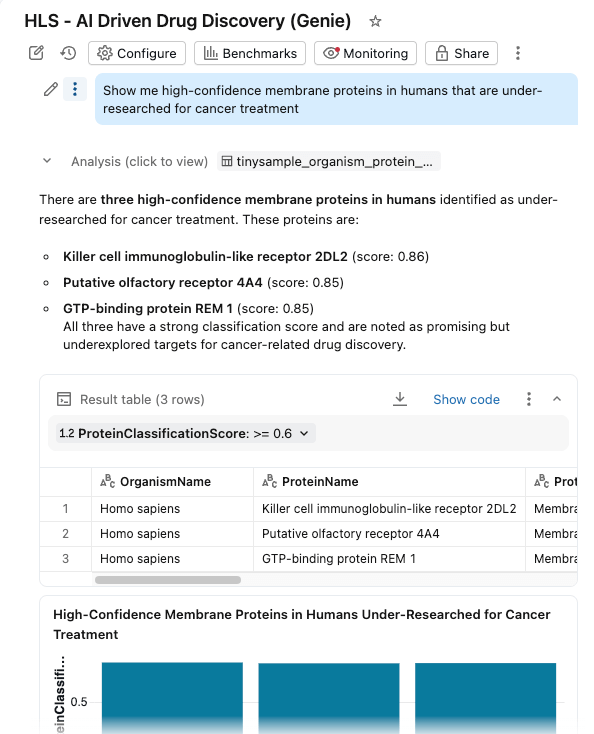
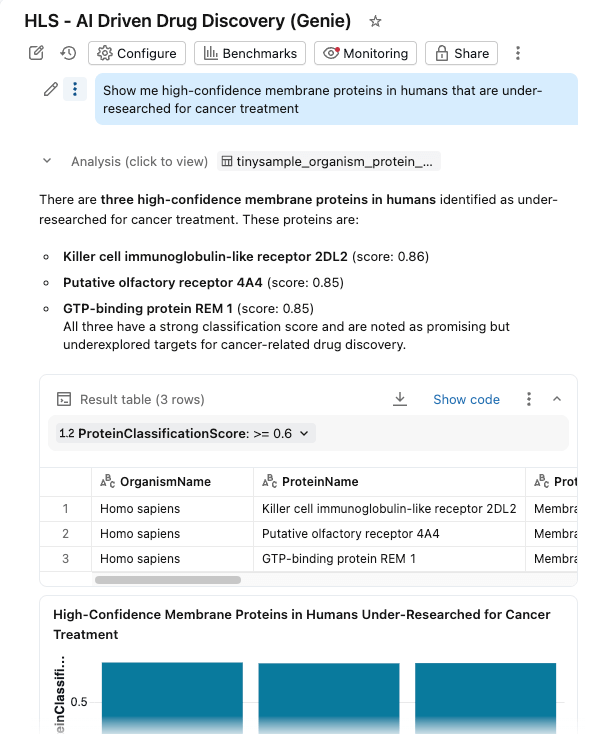{kind=link}
Das Dashboard fragt dieselben verwalteten Tabellen in Unity Catalog ab, wobei KI-Funktionen eine bedarfsgerechte (oder stapelverarbeitete) Anreicherung bereitstellen.
Die Stärke einer einheitlichen Plattform
Was diese Lösung so überzeugend macht, liegt nicht an einer einzelnen Komponente – sondern daran, dass alles auf einer einzigen Plattform läuft:
| Fähigkeit | Databricks-Feature |
|---|---|
| Datenaufnahme & ETL | Lakeflow Declarative Pipelines |
| Data Governance | Unity Catalog |
| ML-Inferenz | GPU Compute |
| LLM-Integration | FMAPI + Externe Modelle + AI Functions |
| Analysen | Databricks SQL |
| Exploration | KI/BI-Dashboards + KI/BI Genie Space |
Entscheidend ist, dass es keine Datenbewegung zwischen den Systemen gibt. Keine separate MLOps-Infrastruktur. Keine getrennten BI-Tools. Die Proteinsequenz, die in die Pipeline gelangt, durchläuft Transformation, Klassifizierung und Anreicherung und ist am Ende in natürlicher Sprache abfragbar – und das alles in derselben kontrollierten Umgebung.
Der vollständige Lösungsbeschleuniger ist auf GitHub verfügbar:
github.com/databricks-industry-solutions/ai-driven-drug-discovery
Was kommt als Nächstes?
Dieser Accelerator zeigt, was alles möglich ist. Im Produktivbetrieb könnten Sie es erweitern, um:
- Verarbeiten Sie die gesamte UniProt-Datenbank mit bereitgestellten Throughput-Endpunkten.
- Weitere (offene oder benutzerdefinierte) Klassifikationsmodelle für verschiedene Proteineigenschaften hinzufügen
- Erstellen Sie RAG -Pipelines für wissenschaftliche Literatur, um fundiertere LLM-Antworten zu erhalten
- Integration in nachgelagerte Workflows für die Molekularsimulation
- Verbindung mit der Proteinstrukturvorhersage (AlphaFold/ESMFold), um klassifizierten Proteinen einen strukturellen 3D-Kontext hinzuzufügen.
- Erweiterung auf andere genomische Formate (FASTQ, VCF, BAM) mit Glow für groß angelegte Sequenzierung und Variantenanalysen
Die Grundlage ist vorhanden. Die Plattform ist vereinheitlicht. Die einzige Grenze ist die Wissenschaft, die Sie beschleunigen möchten. Warum noch warten?
(Dieser Blogbeitrag wurde mit KI-gestützten Tools übersetzt.) Originalbeitrag
Erhalten Sie die neuesten Beiträge in Ihrem Posteingang
Abonnieren Sie unseren Blog und erhalten Sie die neuesten Beiträge direkt in Ihren Posteingang.