Semplificazione delle pipeline genomiche su larga scala con Databricks Delta
Ottieni un'anteprima del nuovo e-book di O'Reilly per la guida dettagliata di cui hai bisogno per iniziare a usare Delta Lake.
Prova questo Notebook in Databricks
Questo è il primo blog della nostra serie "Analisi genomica su vasta scala". In questa serie, dimostreremo come la Piattaforma di analisi unificata per la genomica di Databricks consenta ai clienti di analizzare dati genomici su scala di popolazione. Partendo dall'output della nostra pipeline genomica, questa serie fornirà un tutorial sull'uso di Databricks per eseguire il controllo qualità dei campioni, la genotipizzazione congiunta, il controllo qualità della coorte e analisi avanzate di genetica statistica.
Dal completamento del Progetto Genoma Umano nel 2003, si è assistito a un'esplosione di dati alimentata da un drastico calo del costo del sequenziamento del DNA, dai 3 miliardi di dollari1 per il primo genoma a meno di 1.000 dollari di oggi.
[1] Il Progetto Genoma Umano è stato un progetto da 3 miliardi di dollari guidato dal Dipartimento dell'Energia e dai National Institutes of Health, iniziato nel 1990 e completato nel 2003.
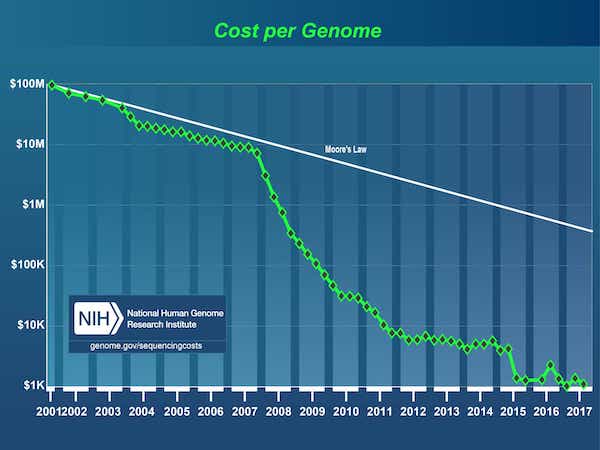
Fonte: Costi del sequenziamento del DNA: Dati
Di conseguenza, il campo della genomica è ora maturato fino a un punto in cui le aziende hanno iniziato a eseguire il sequenziamento del DNA su scala di popolazione. Tuttavia, il sequenziamento del codice del DNA è solo il primo passo: i dati grezzi devono poi essere trasformati in un formato adatto all'analisi. Generalmente, questo viene fatto unendo una serie di strumenti di bioinformatica con script personalizzati ed elaborando i dati su un singolo nodo, un campione alla volta, fino a ottenere una raccolta di varianti genomiche. Gli scienziati di Bioinformatica oggi trascorrono la maggior parte del loro tempo a creare e manutenere queste pipeline. Dato che i set di dati genomici hanno raggiunto la scala dei petabyte, è diventato difficile rispondere tempestivamente anche alle seguenti semplici domande:
- Quanti campioni abbiamo sequenziato questo mese?
- Qual è il numero totale di varianti uniche rilevate?
- Quante varianti abbiamo osservato nelle diverse classi di variazione?
A complicare ulteriormente questo problema, i dati di migliaia di individui non possono essere archiviati, tracciati o versionati pur rimanendo accessibili e interrogabili. Di conseguenza, i ricercatori spesso duplicano sottoinsiemi dei loro dati genomici durante le analisi, causando un aumento dell'ingombro di archiviazione e dei costi complessivi. Nel tentativo di alleviare questo problema, oggi i ricercatori impiegano una strategia di "congelamento dei dati" (data freeze), in genere per un periodo da sei mesi a due anni, durante il quale interrompono il lavoro su nuovi dati e si concentrano invece su una copia congelata dei dati esistenti. Non esiste una soluzione per sviluppare in modo incrementale le analisi in tempi più brevi, causando un rallentamento del progresso della ricerca.
C'è una forte necessità di un software robusto in grado di elaborare dati genomici su scala industriale, mantenendo al contempo la flessibilità per gli scienziati di esplorare i dati, iterare sulle loro pipeline di analisi e ricavare nuove conoscenze.
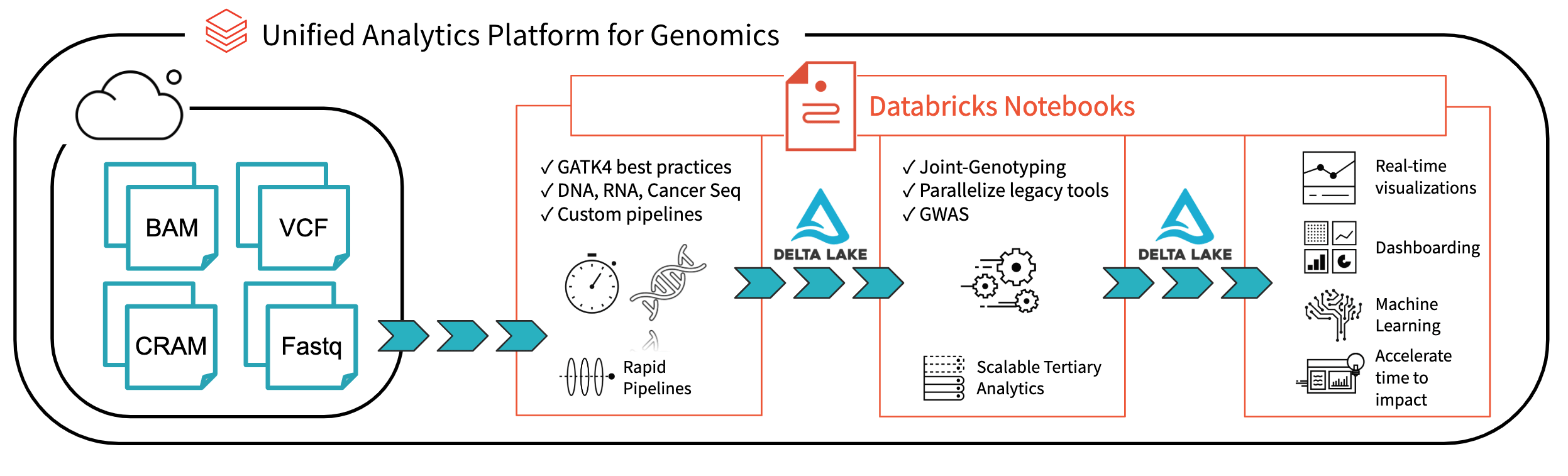
Fig. 1. Architettura per l'analisi genomica end-to-end con Databricks
Con Databricks Delta: un sistema di gestione unificato per l'analisi dei Big Data in tempo reale, la piattaforma Databricks ha compiuto un passo importante verso la risoluzione dei problemi di governance dei dati, accesso e analisi dei dati che i ricercatori devono affrontare oggi. Con Databricks Delta Lake, puoi archiviare tutti i tuoi dati genomici in un unico posto e creare analisi che si aggiornano in tempo reale man mano che nuovi dati vengono inseriti. In combinazione con le ottimizzazioni della nostra Piattaforma di analisi unificata per la genomica (UAP4G) per la lettura, la scrittura e l'elaborazione dei formati di file genomici, offriamo una soluzione end-to-end per i flussi di lavoro delle pipeline genomiche. L'architettura UAP4G offre flessibilità, consentendo ai clienti di integrare le proprie pipeline e sviluppare le proprie analitiche terziarie. Ad esempio, abbiamo evidenziato la dashboard seguente che mostra metriche e visualizzazioni di controllo qualità che possono essere calcolate e presentate in modo automatico e personalizzate per soddisfare requisiti specifici.
https://www.youtube.com/watch?v=73fMhDKXykU
Nel resto di questo blog, illustreremo i passaggi che abbiamo seguito per creare la dashboard di controllo qualità riportata sopra, che si aggiorna in tempo reale man mano che l'elaborazione dei campioni viene completata. Utilizzando una pipeline basata su Delta per l'elaborazione dei dati genomici, i nostri clienti possono ora gestire le loro pipeline in modo da fornire visibilità in tempo reale, campione per campione. Con i notebook Databricks (e integrazioni come GitHub e MLflow) possono tracciare e versionare le analisi in modo da garantire la riproducibilità dei risultati. I loro bioinformatici possono dedicare meno tempo alla manutenzione delle pipeline e più tempo a fare scoperte. Consideriamo l'UAP4G come il motore che guiderà la trasformazione dalle analisi ad hoc alla genomica di produzione su scala industriale, consentendo di ottenere approfondimenti migliori sul link tra genetica e malattia.
Leggi dati di esempio
Iniziamo leggendo i dati di variazione da una piccola coorte di campioni; l'istruzione seguente legge i dati per un sampleId specifico e li salva utilizzando il formato Databricks Delta (nella cartella delta_stream_output).
Nota: la cartella annotations_etl_parquet contiene annotazioni generate dal set di dati 1000 Genomes archiviato in formato Parquet. L'ETL e l'elaborazione di queste annotazioni sono stati eseguiti utilizzando Databricks’ Piattaforma di analisi unificata per la genomica.
Start lo streaming della tabella Databricks Delta
Nella seguente istruzione, stiamo creando il DataFrame Apache Spark exomes, che legge un stream (tramite readStream) di dati utilizzando il formato Databricks Delta. Si tratta di un DataFrame a esecuzione continua o dinamico, ovvero il DataFrame exomes caricherà nuovi dati man mano che vengono scritti nella cartella delta_stream_output. Per visualizzare il DataFrame exomes, possiamo eseguire una query sul DataFrame per trovare il conteggio delle varianti raggruppate per sampleId.
Quando si esegue l'istruzione display, il notebook di Databricks fornisce una dashboard di streaming per monitorare i job di streaming. Subito sotto il job di streaming si trovano i risultati dell'istruzione display (ossia il conteggio delle varianti per sample_id).
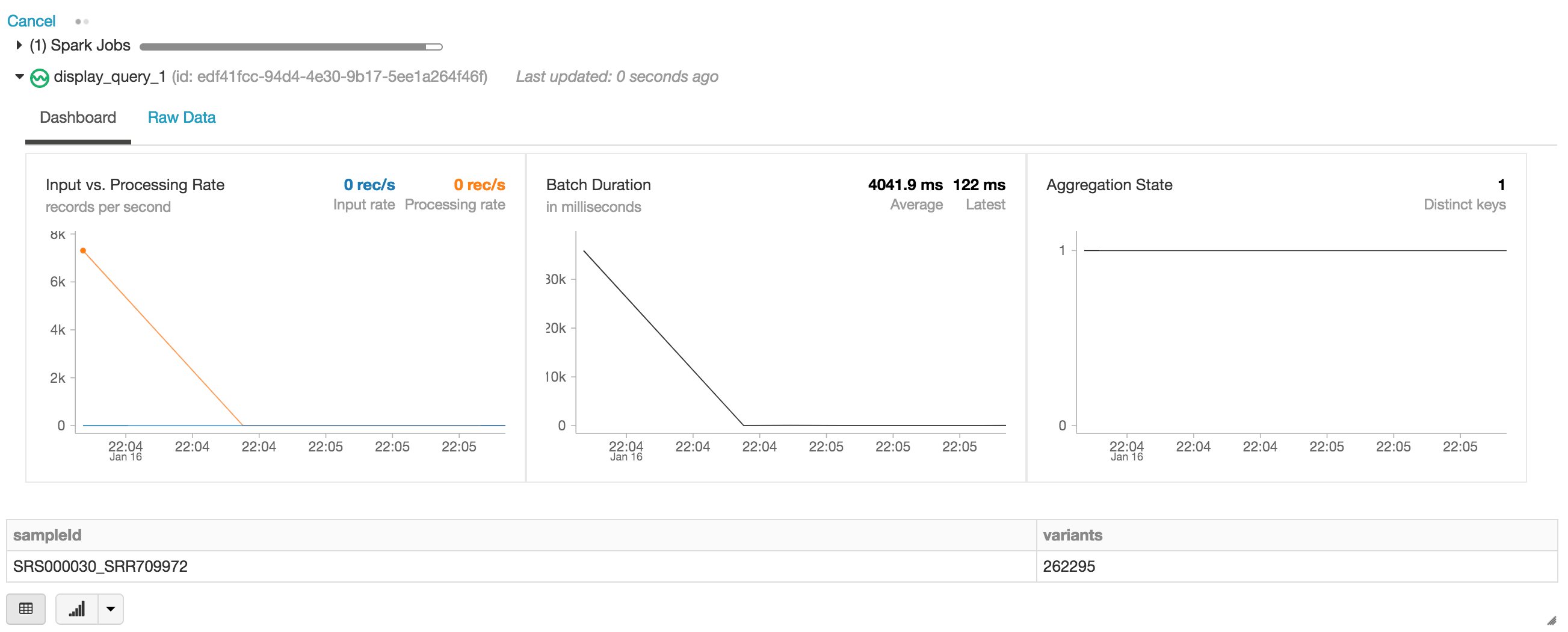
Continuiamo a rispondere alle domande iniziali eseguendo altre query basate sul nostro DataFrame `exomes`.
Conteggio delle varianti a singolo nucleotide
Per continuare con l'esempio, possiamo calcolare rapidamente il numero di varianti a singolo nucleotide (SNV), come mostrato nel graph seguente.
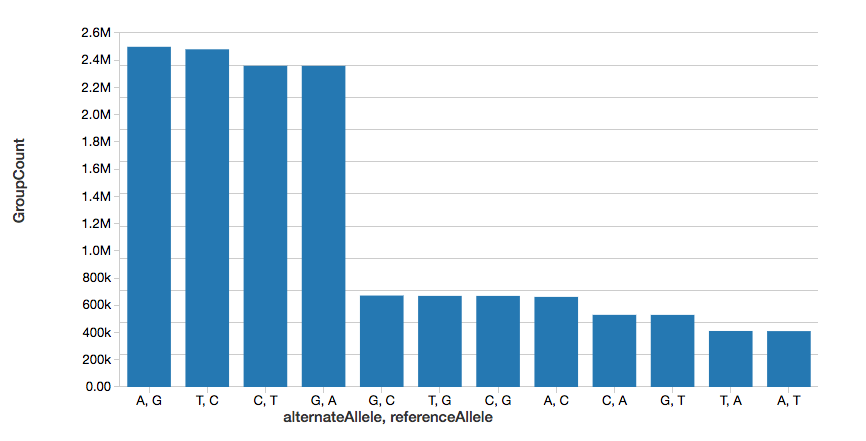
Nota: il comando display fa parte del workspace di Databricks che consente di visualizzare il DataFrame utilizzando le visualizzazioni di Databricks (ad es. nessuna codifica richiesta).
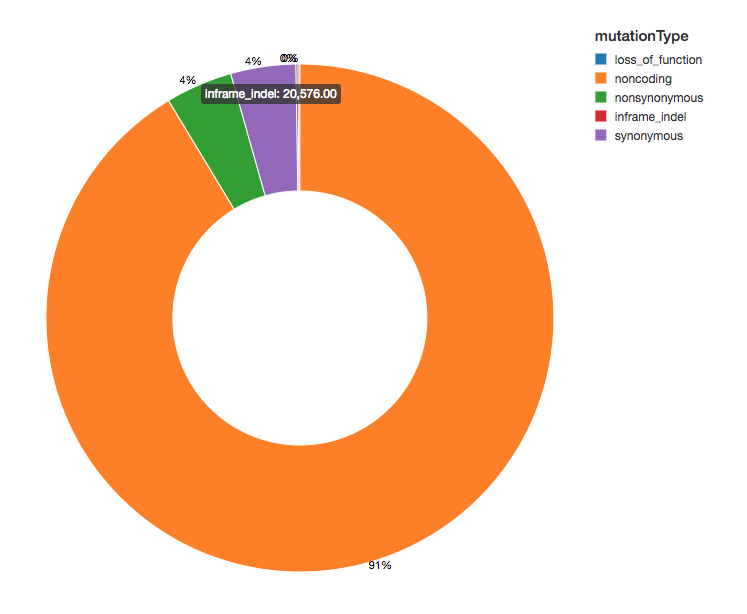
Conteggio delle varianti
Poiché abbiamo annotato le nostre varianti con effetti funzionali, possiamo proseguire la nostra analisi esaminando la distribuzione degli effetti delle varianti che osserviamo. La maggior parte delle varianti rilevate fiancheggia le regioni che codificano per le proteine; queste sono note come varianti non codificanti.
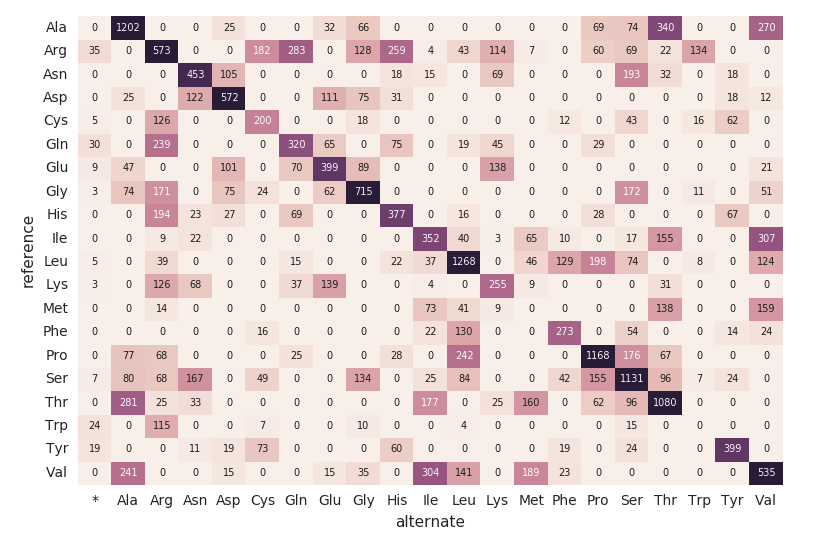
Mappa di calore della sostituzione degli aminoacidi
Proseguendo con il nostro DataFrame exomes, calcoliamo i conteggi delle sostituzioni di amminoacidi con il seguente snippet di codice. Analogamente ai DataFrame precedenti, creeremo un altro DataFrame dinamico (aa_counts) in modo tale che, man mano che nuovi dati vengono elaborati dal DataFrame degli esomi, ciò si rifletta di conseguenza anche nei conteggi delle sostituzioni di amminoacidi. Stiamo anche scrivendo i dati in memoria (ad es. .format("memory")) e batch elaborati ogni 60s (ossia trigger(processingTime=’60 seconds’)) in modo che il codice della heatmap di Pandas a valle possa elaborare e visualizzare la heatmap.
Il seguente frammento di codice legge la precedente tabella Spark amino_acid_substitutions, determina il conteggio massimo, crea una nuova tabella pivot Pandas dalla tabella Spark, quindi traccia la mappa di calore.
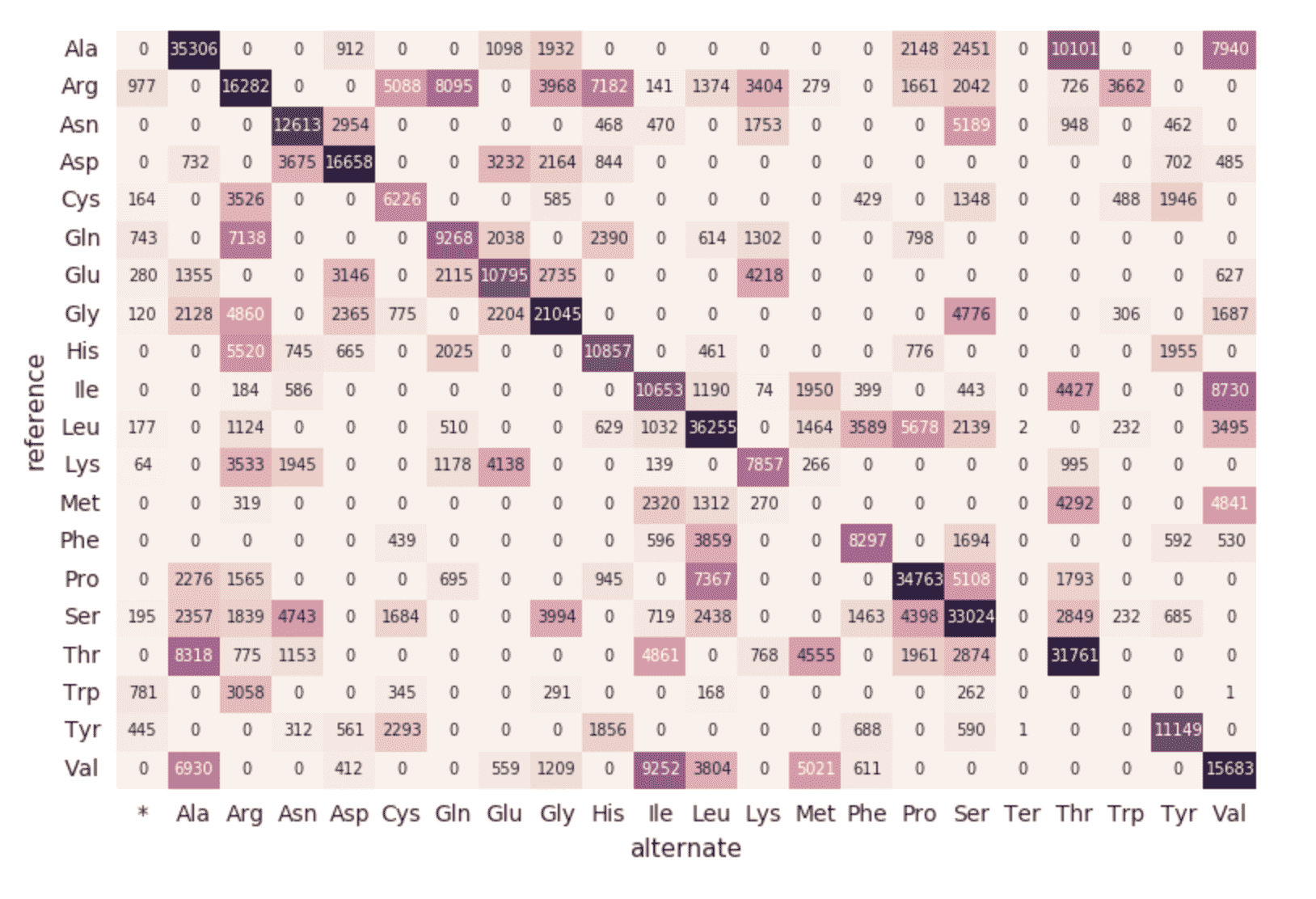
Migrazione a una pipeline continua
Fino a questo punto, gli snippet di codice e le visualizzazioni precedenti rappresentano una singola esecuzione per un singolo sampleId. Ma poiché stiamo usando Structured Streaming e Databricks Delta, questo codice può essere usato (senza alcuna modifica) per costruire una pipeline di dati di produzione che calcola continuamente le statistiche di controllo qualità man mano che i campioni passano attraverso la nostra pipeline. Per dimostrarlo, possiamo eseguire il seguente snippet di codice che caricherà il nostro intero set di dati.
Come descritto nei frammenti di codice precedenti, l'origine del DataFrame exomes sono i file caricati nella cartella delta_stream_output. Inizialmente, avevamo caricato un set di file per un singolo sampleId (ad esempio, sampleId = “SRS000030_SRR709972”). Lo snippet di codice precedente ora acquisisce tutti i campioni parquet generati (ossia parquets) e carica in modo incrementale tali file per sampleId nella stessa cartella delta_stream_output. La seguente GIF animata mostra l'output abbreviato dello snippet di codice precedente.
https://www.youtube.com/watch?v=JPngSC5Md-Q
Visualizzazione della pipeline genomica
Quando torni all'inizio del notebook, noterai che il DataFrame exomes sta caricando automaticamente i nuovi sampleIds. Poiché il componente di streaming strutturato della nostra pipeline genomica viene eseguito in modo continuo, elabora i dati non appena i nuovi file vengono caricati nella cartella delta_stream_outputpath. Utilizzando il formato Databricks Delta, possiamo garantire la coerenza transazionale dei dati trasmessi in streaming al DataFrame exomes.
https://www.youtube.com/watch?v=Q7KdPsc5mbY
A differenza della creazione iniziale del nostro DataFrame exomes, si può notare che il dashboard di monitoraggio dello streaming strutturato sta caricando i dati (ossia la fluttuazione della "velocità di input rispetto a quella di elaborazione", la fluttuazione della "durata del batch" e un aumento delle chiavi distinte nello "stato delle aggregazioni"). Mentre il DataFrame exomes è in fase di elaborazione, notare le nuove righe di sampleIds (e i conteggi delle varianti). Questa stessa azione si può osservare anche per la query associata raggruppata per tipo di mutazione.
https://www.youtube.com/watch?v=sT179SCknGM
Con Databricks Delta, qualsiasi nuovo dato è transazionalmente coerente in ogni singola fase della nostra pipeline genomica. Ciò è importante perché garantisce che la pipeline sia coerente (mantiene la coerenza dei dati, ovvero garantisce che tutti i dati siano “corretti”), affidabile (la transazione riesce o fallisce completamente) e in grado di gestire aggiornamenti in tempo reale (la capacità di gestire molte transazioni contemporaneamente e qualsiasi interruzione o errore non avrà alcun impatto sui dati). Pertanto, anche i dati nella nostra mappa di sostituzione degli amminoacidi downstream (che ha richiesto una serie di passaggi ETL aggiuntivi) vengono aggiornati senza soluzione di continuità.
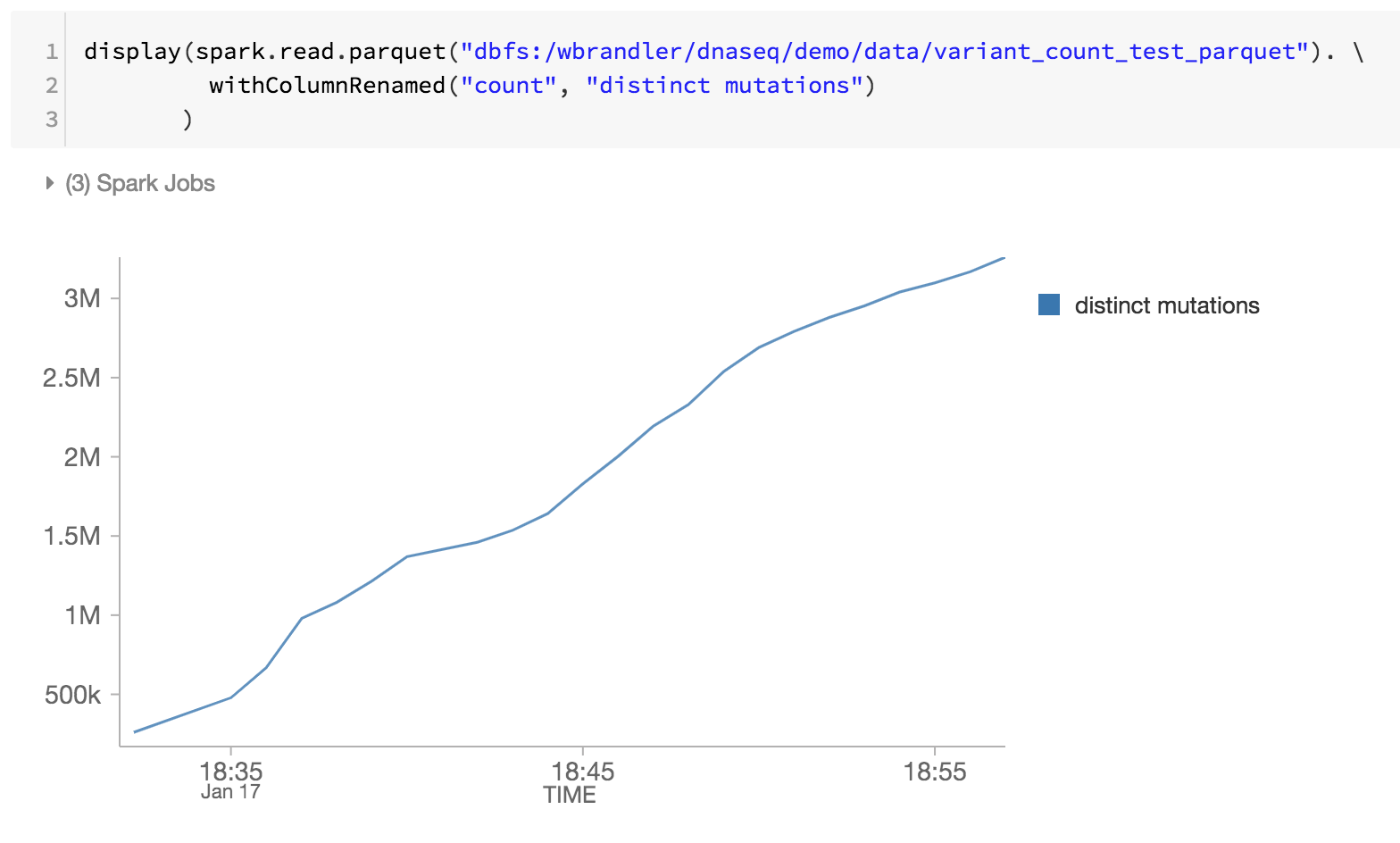
Come ultimo passaggio della nostra pipeline genomica, stiamo anche monitorando le mutazioni distinte esaminando i file Parquet di Databricks Delta all'interno di DBFS (ovvero l'aumento di mutazioni distinte nel tempo).
Riepilogo
Utilizzando le fondamenta della Databricks Piattaforma di analisi unificata, con un'attenzione particolare a Databricks Delta, bioinformatici e ricercatori possono applicare le analitiche distribuite con coerenza transazionale usando Databricks Piattaforma di analisi unificata per la genomica. Queste astrazioni consentono ai professionisti dei dati di semplificare le pipeline genomiche. Qui abbiamo creato una pipeline di controllo qualità per campioni genomici che elabora continuamente i dati man mano che vengono processati nuovi campioni, senza intervento manuale. Sia che si esegua l'ETL o si eseguano analitiche sofisticate, i dati fluiranno attraverso la pipeline genomica rapidamente e senza interruzioni. Provalo oggi stesso scaricando il Notebook Semplificare le pipeline genomiche su larga scala con Databricks Delta.
Inizia ad analizzare la genomica su larga scala:
- Leggi la nostra guida alla soluzioneUnified Analytics per la genomica
- Download the Simplifying genomica pipeline at Scale with Databricks Delta Notebook
- Iscriviti a una prova gratuita di Databricks Unified Analytics for Genomics
Ringraziamenti
Grazie a Yongsheng Huang e Michael Ortega per i loro contributi.
Visita l'hub online di Delta Lake per saperne di più, scaricare il codice più recente e unirti alla community di Delta Lake.
Ricevi gli ultimi articoli nella tua casella di posta
Iscriviti al nostro blog e ricevi gli ultimi articoli direttamente nella tua casella di posta.