Simplifying Genomics Pipelines at Scale with Databricks Delta
von William Brandler und Frank Austin Nothaft
Holen Sie sich eine Vorschau auf das neue E-Book von O'Reilly für die Schritt-für-Schritt-Anleitung, die Sie für den Einstieg in Delta Lake benötigen.
Testen Sie dieses Notebook in Databricks
Dieser Blogbeitrag ist der erste in unserer Reihe „Genomik Analysen at Scale“. In dieser Reihe zeigen wir, wie die Databricks einheitliche Analyseplattform für Genomik es Kunden ermöglicht, Genomikdaten im Populationsmaßstab zu analysieren. Ausgehend von der Ausgabe unserer Genomik-Pipeline bietet diese Reihe ein Tutorial zur Ausführung von Databricks zur Ausführung von Probenqualitätskontrollen, gemeinsamen Genotypisierungen, Kohortenqualitätskontrollen und erweiterten statistischen genetischen Analysen.
Seit dem Abschluss des Humangenomprojekts im Jahr 2003 gab es eine Datenexplosion, die durch einen drastischen Preisverfall bei der DNA-Sequenzierung befeuert wurde: von 3 Mrd. $1 für das erste Genom auf heute unter 1.000 $.
[1] Das Humangenomprojekt war ein 3-Milliarden-Dollar-Projekt unter der Leitung des Department of Energy und der National Institutes of Health, das 1990 begann und 2003 abgeschlossen wurde.
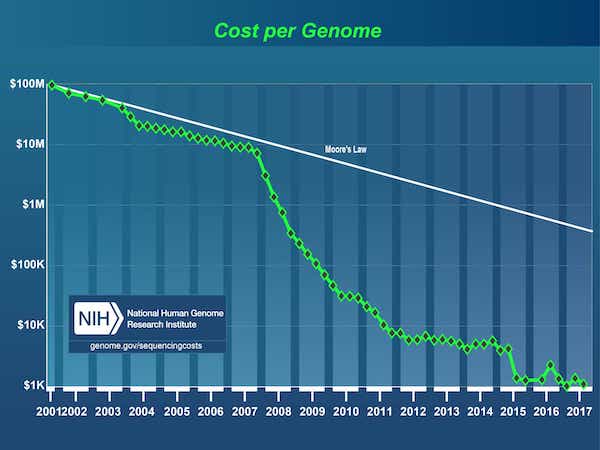
Quelle: Kosten der DNA-Sequenzierung: Daten
Daher ist der Bereich der Genomik mittlerweile so weit fortgeschritten, dass Unternehmen begonnen haben, DNA-Sequenzierungen im Bevölkerungs-Scale durchzuführen. Die Sequenzierung des DNA-Codes ist jedoch nur der erste Schritt. Anschließend müssen die Rohdaten in ein für die Analyse geeignetes Format umgewandelt werden. Typischerweise geschieht dies durch das Zusammenfügen einer Reihe von Bioinformatik -Tools mit benutzerdefinierten Skripten und die Verarbeitung der Daten auf einem einzigen Knoten, eine Probe nach der anderen, bis wir schließlich eine Sammlung genomischer Varianten erhalten. Bioinformatiker verbringen heute den größten Teil ihrer Zeit mit dem Aufbau und der Wartung dieser Pipelines. Da genomische Datensätze in den Petabyte-Scale angewachsen sind, ist es zu einer Herausforderung geworden, selbst die folgenden einfachen Fragen zeitnah zu beantworten:
- Wie viele Proben haben wir diesen Monat sequenziert?
- Wie viele eindeutige Varianten wurden erkannt?
- Wie viele Varianten haben wir in den verschiedenen Variationsklassen gesehen?
Dieses Problem wird dadurch verschärft, dass Daten von Tausenden von Personen nicht gespeichert, nachverfolgt oder versioniert werden können und gleichzeitig zugänglich und abfragbar bleiben. Infolgedessen duplizieren Forscher bei der Durchführung ihrer Analysen häufig Teilmengen ihrer genomischen Daten, was den gesamten Speicherbedarf und die Kosten in die Höhe treibt. Um dieses Problem zu lindern, wenden Forscher heute eine Strategie der „Data Freezes“ an: Für einen Zeitraum von typischerweise sechs Monaten bis zu zwei Jahren stellen sie die Arbeit an neuen Daten ein und konzentrieren sich stattdessen auf eine eingefrorene Kopie vorhandener Daten. Es gibt keine Lösung, um Analysen über kürzere Zeiträume inkrementell aufzubauen, was den Forschungsfortschritt verlangsamt.
Es besteht ein dringender Bedarf an robuster Software, die genomische Daten im industriellen Maßstab verarbeiten kann und gleichzeitig Wissenschaftlern die Flexibilität bietet, die Daten zu untersuchen, ihre Analyse-Pipelines zu iterieren und neue Erkenntnisse zu gewinnen.
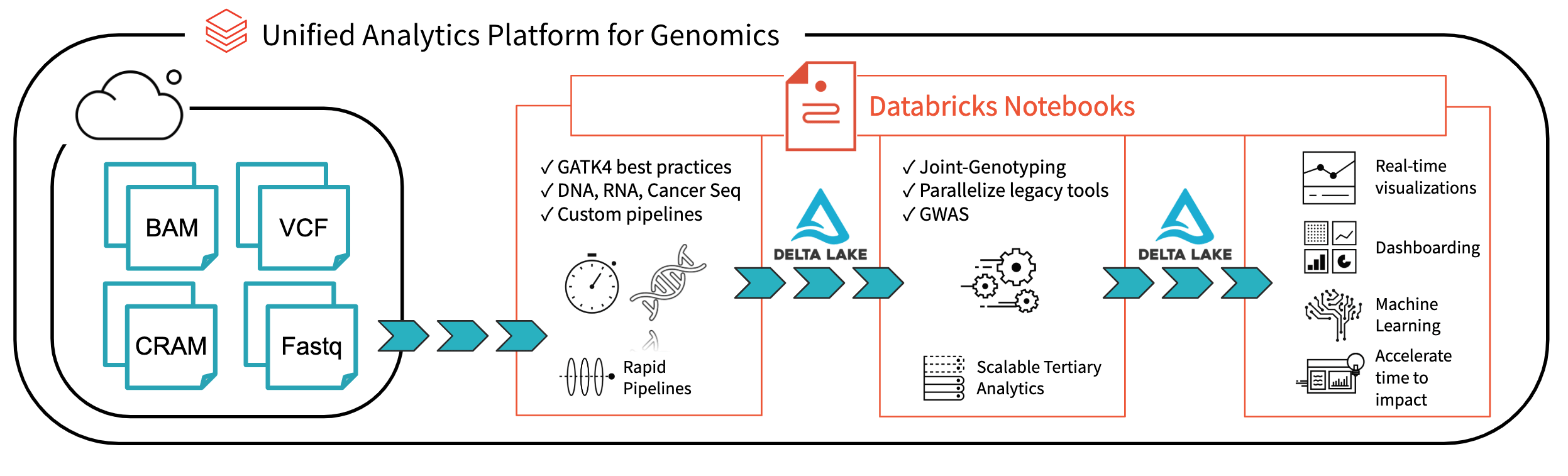
Abb. 1. Architektur für die End-to-End-Genomanalyse mit Databricks
Mit Databricks Delta: A Unified Management System for Echtzeit Big Data Analytics hat die Databricks-Plattform einen großen Schritt zur Lösung der Probleme in den Bereichen Data Governance, Datenzugriff und Datenanalysen gemacht, mit denen Forscher heute konfrontiert sind. Mit Databricks Delta Lake können Sie all Ihre Genomdaten an einem Ort speichern und Analysen erstellen, die in Echtzeit aktualisiert werden, sobald neue Daten aufgenommen werden. In Kombination mit Optimierungen in unserer einheitlichen Analyseplattform für Genomik (UAP4G) zum Lesen, Schreiben und Verarbeiten von Genomik-Dateiformaten bieten wir eine End-to-End-Lösung für die Workflows von Genomik-Pipelines. Die UAP4G-Architektur bietet Flexibilität und ermöglicht es Kunden, ihre eigenen Pipelines anzuschließen und ihre eigenen tertiären Analysen zu entwickeln. Als Beispiel haben wir das folgende Dashboard hervorgehoben, das Metriken zur Qualitätskontrolle und Visualisierungen zeigt, die automatisiert berechnet, dargestellt und an Ihre spezifischen Anforderungen angepasst werden können.
https://www.youtube.com/watch?v=73fMhDKXykU
Im weiteren Verlauf dieses Blogs führen wir Sie durch die Schritte zur Erstellung des obigen Dashboards für die Qualitätskontrolle, das in Echtzeit aktualisiert wird, sobald die Verarbeitung von Samples abgeschlossen ist. Durch die Verwendung einer Delta-basierten Pipeline für die Verarbeitung genomischer Daten können unsere Kunden ihre Pipelines nun so betreiben, dass eine Echtzeit-Transparenz auf Stichprobenebene gewährleistet ist. Mit Databricks-Notebooks (und Integrationen wie GitHub und MLflow) können sie Analysen so nachverfolgen und versionieren, dass die Reproduzierbarkeit ihrer Ergebnisse gewährleistet ist. Ihre Bioinformatiker können weniger Zeit für die Wartung von Pipelines aufwenden und mehr Zeit für Entdeckungen nutzen. Wir sehen das UAP4G als den Motor, der die Transformation von Ad-hoc-Analysen zur Produktionsgenomik im industriellen Maßstab vorantreiben und so bessere Einblicke in den Zusammenhang zwischen Genetik und Krankheit ermöglichen wird.
Beispieldaten lesen
Starten wir mit dem Einlesen von Variationsdaten aus einer kleinen Kohorte von Proben. Die folgende Anweisung liest Daten für eine bestimmte sampleId ein und speichert sie im Databricks-Delta-Format (im Ordner delta_stream_output).[[ ## completed ## ]]
Hinweis: Der Ordner „annotations_etl_parquet“ enthält Annotationen, die aus dem 1000-Genome-Dataset generiert und im Parquet-Format gespeichert wurden. Das ETL und die Verarbeitung dieser Annotationen erfolgte über die einheitliche Analyseplattform von Databricks für Genomik.
Streaming der Databricks Delta-Tabelle starten
In der folgenden Anweisung erstellen wir das Exomes Apache Spark DataFrame, das einen stream (über readStream) von Daten im Databricks Delta-Format liest. Dies ist ein kontinuierlich ausgeführter oder dynamischer DataFrame, d. h. der Exomes-DataFrame lädt neue Daten, sobald diese in den Ordner 'delta_stream_output' geschrieben werden. Um den Exomes-DataFrame anzuzeigen, können wir eine DataFrame-Abfrage ausführen, um die Anzahl der nach der sampleId gruppierten Varianten zu ermitteln.
Bei der Ausführung der display -Anweisung stellt das Databricks-Notebook ein Streaming-Dashboard zur Überwachung der Streaming-Jobs bereit. Direkt unter dem Streaming-Job befinden sich die Ergebnisse der display-Anweisung (d. h. die Anzahl der Varianten nach sample_id).
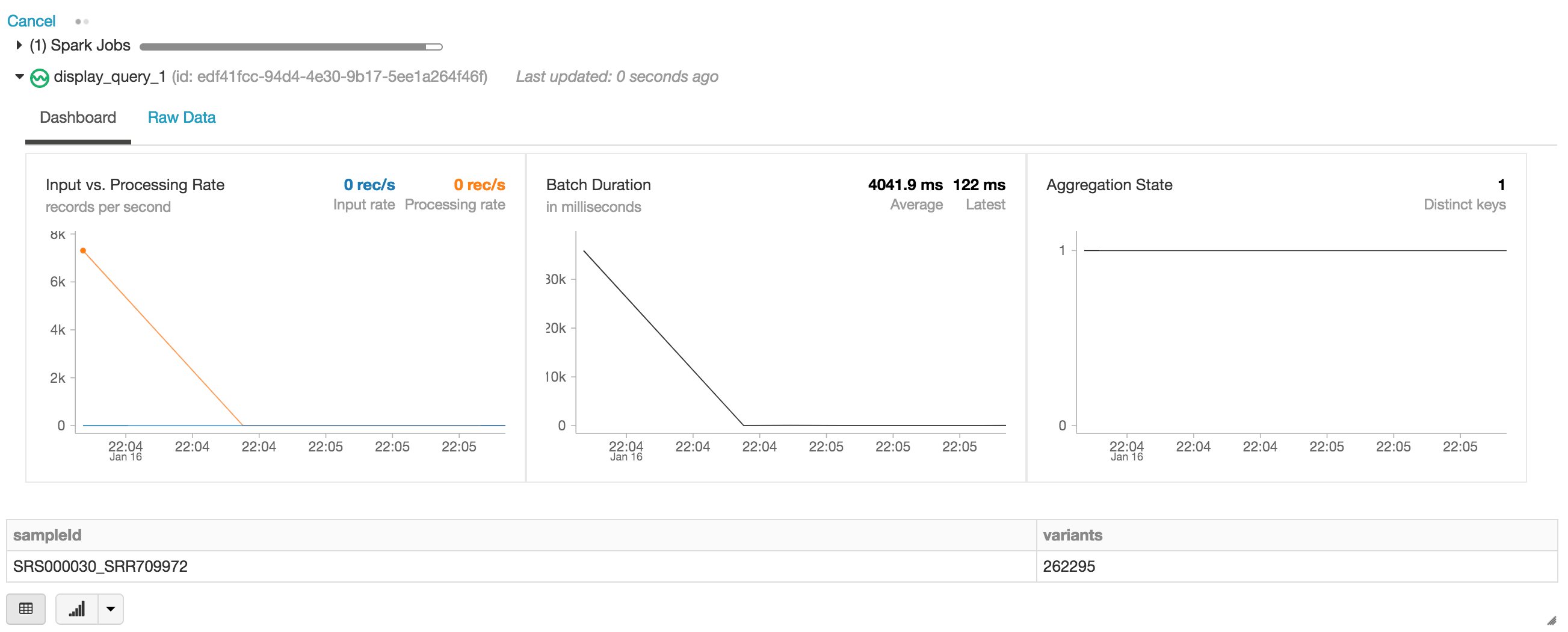
Beantworten wir unsere anfänglichen Fragen, indem wir weitere DataFrame-Abfragen auf der Grundlage unseres exomes -DataFrames ausführen.
Anzahl der Einzelnukleotidvarianten
Um das Beispiel fortzusetzen, können wir schnell die Anzahl der Einzelnukleotid-Varianten (SNVs) berechnen, wie in dem folgenden Graph dargestellt.
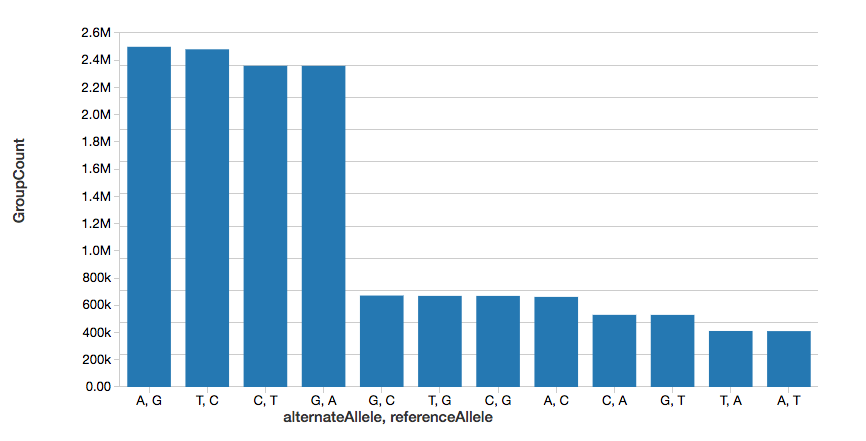
Beachten Sie, dass der Befehl display Teil des Databricks-Workspace ist, mit dem Sie Ihren DataFrame mithilfe von Databricks-Visualisierungen anzeigen können (d. h. kein Code erforderlich).
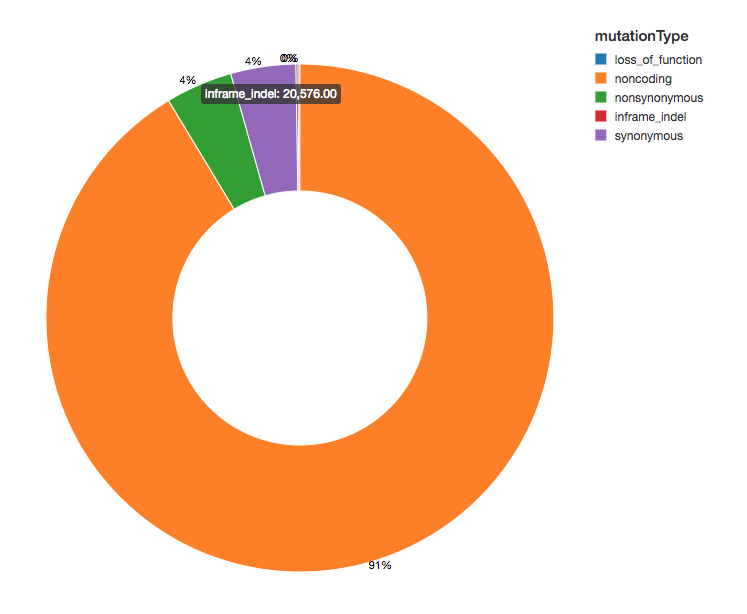
Anzahl der Varianten
Da wir unsere Varianten mit funktionellen Auswirkungen annotiert haben, können wir unsere Analysen fortsetzen, indem wir uns die Verteilung der beobachteten Varianten-Auswirkungen ansehen. Die meisten der entdeckten Varianten flankieren Regionen, die für Proteine kodieren, diese sind als nicht kodierende Varianten bekannt.
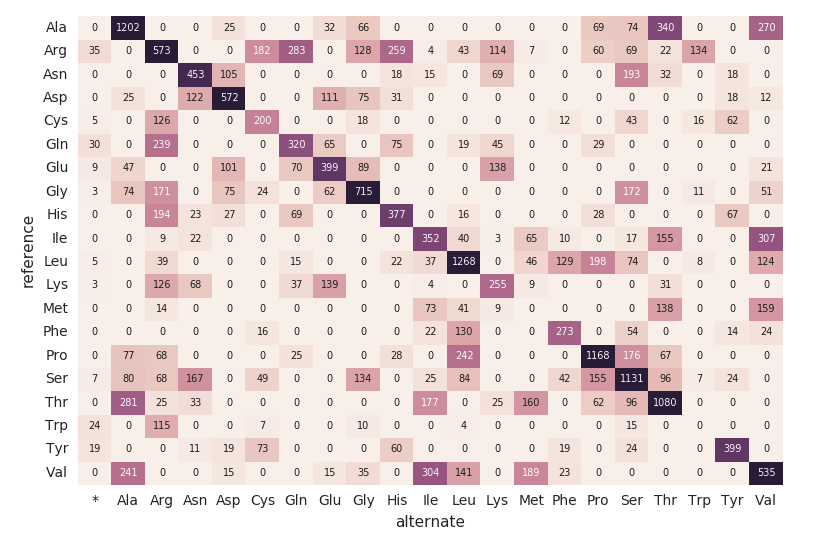
Aminosäure-Substitutions-Heatmap
Fahren wir mit unserem exomes -DataFrame fort und berechnen wir die Anzahl der Aminosäuresubstitutionen mit dem folgenden Code-Snippet. Ähnlich wie bei den vorherigen DataFrames erstellen wir einen weiteren dynamischen DataFrame (aa_counts), sodass, wenn neue Daten vom exomes-DataFrame verarbeitet werden, sich dies anschließend auch in der Anzahl der Aminosäuresubstitutionen widerspiegelt. Wir schreiben die Daten auch in den Arbeitsspeicher (d. h. .format(“memory”)) und verarbeiten Batches alle 60 Sekunden (d. h. trigger(processingTime=’60 seconds’)), damit der nachgelagerte Pandas-Heatmap-Code die Heatmap verarbeiten und visualisieren kann.
Der folgende Codeausschnitt liest die vorangehende amino_acid_substitutions -Spark-Tabelle, bestimmt die maximale Anzahl, erstellt eine neue Pandas-Pivot-Tabelle aus der Spark-Tabelle und zeichnet dann die Heatmap.
Migration zu einer kontinuierlichen Pipeline
Bisher stellen die vorangegangenen Code-Snippets und Visualisierungen eine einzelne Ausführung für eine einzelne sampleId dar. Da wir jedoch Structured Streaming und Databricks Delta verwenden, kann dieser Code (ohne Änderungen) verwendet werden, um eine Produktions-Datenpipeline zu erstellen, die kontinuierlich Statistiken zur Qualitätskontrolle berechnet, während Samples unsere Pipeline durchlaufen. Um dies zu demonstrieren, können wir das folgende Code-Snippet ausführen, das unser gesamtes Dataset lädt.
Wie in den vorherigen Code-Snippets beschrieben, sind die in den Ordner delta_stream_output geladenen Dateien die Quelle des exomes -DataFrame. Ursprünglich hatten wir einen Satz Dateien für eine einzelne sampleId (d. h. sampleId = “SRS000030_SRR709972”) geladen. Der vorhergehende Code-Snippet nimmt jetzt alle generierten Parquet-Samples (d. h. Parquets) und lädt diese Dateien inkrementell nach sampleId in denselben Ordner delta_stream_output. Die folgende animierte GIF zeigt die gekürzte Ausgabe des vorhergehenden Code-Snippets.
https://www.youtube.com/watch?v=JPngSC5Md-Q
Visualisierung Ihrer Genomik-Pipeline
Wenn Sie zum Anfang Ihres Notebooks zurückscrollen, werden Sie feststellen, dass der exomes -DataFrame jetzt automatisch die neuen sampleIds lädt. Da die Komponente für strukturiertes Streaming unserer Genomik-Pipeline kontinuierlich ausgeführt wird, verarbeitet sie Daten, sobald neue Dateien in den Ordner delta_stream_outputpath geladen werden. Durch die Verwendung des Databricks-Delta-Formats können wir die transaktionale Konsistenz der Daten gewährleisten, die in das Exome-DataFrame gestreamt werden.
https://www.youtube.com/watch?v=Q7KdPsc5mbY
Im Gegensatz zur anfänglichen Erstellung unseres Exome-DataFrame kannst du sehen, wie das strukturiertes Streaming-Monitoring-Dashboard jetzt Daten lädt (d. h. die schwankende "Eingabe- vs. Verarbeitungsrate", die schwankende "Batch-Laufzeit" und eine Zunahme der verschiedenen Keys im "Aggregationsstatus"). Beachten Sie während der Verarbeitung des exomes -DataFrames die neuen Zeilen von sampleIds (und die Anzahl der Varianten). Dieselbe Aktion ist auch bei der Abfrage der zugehörigen Gruppe nach Mutationsart zu sehen.
https://www.youtube.com/watch?v=sT179SCknGM
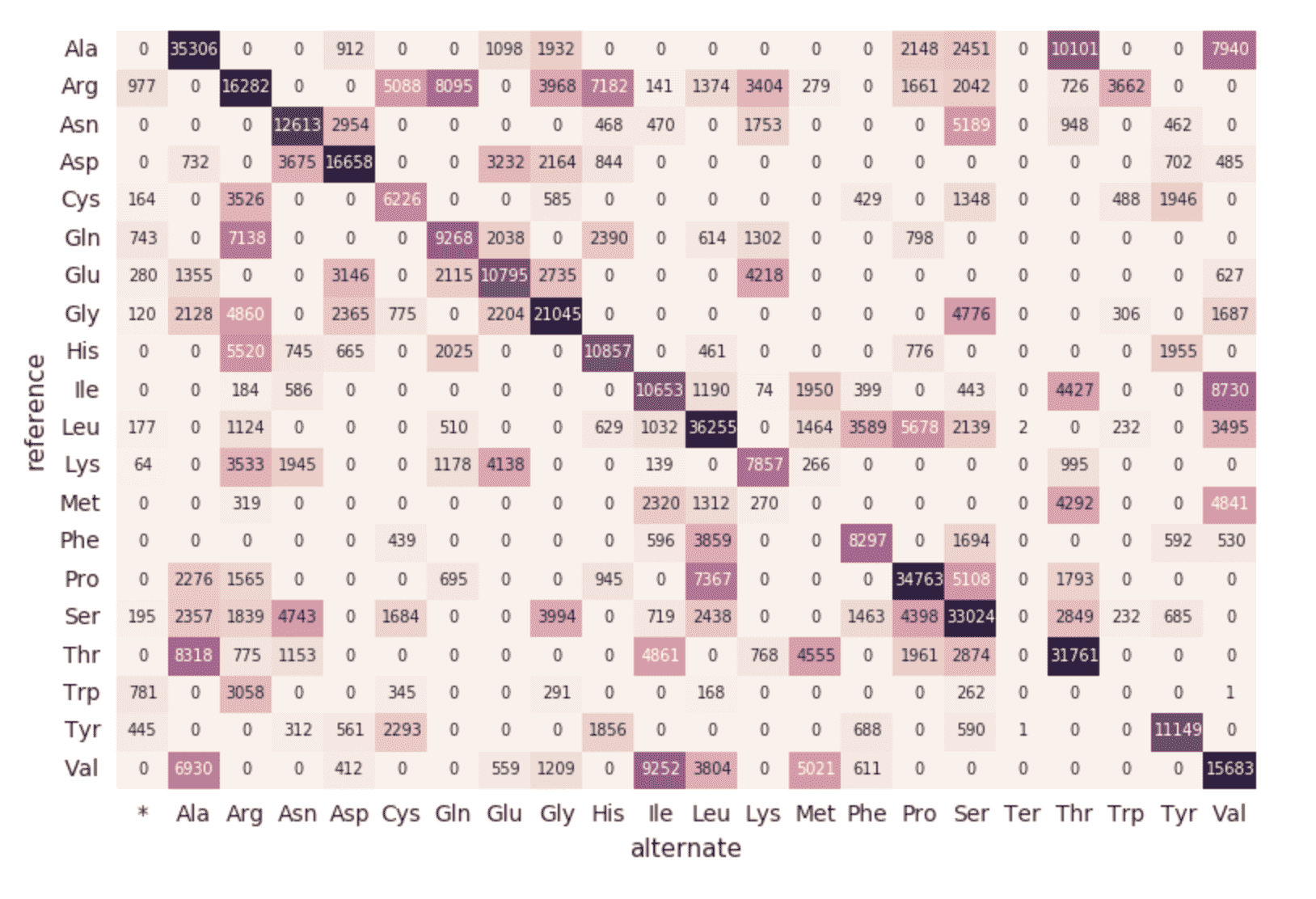
Mit Databricks Delta sind alle neuen Daten in jedem einzelnen Schritt unserer Genomik-Pipeline transaktional konsistent. Dies ist wichtig, da dadurch sichergestellt wird, dass Ihre Pipeline konsistent ist (die Konsistenz Ihrer Daten aufrechterhält, d. h. sicherstellt, dass alle Daten „korrekt“ sind), zuverlässig ist (entweder die Transaktion erfolgreich ist oder vollständig fehlschlägt) und Echtzeit-Updates verarbeiten kann (die Fähigkeit, viele Transaktionen gleichzeitig zu verarbeiten, und jeder Ausfall die Daten nicht beeinträchtigt). Daher werden sogar die Daten in unserer nachgelagerten Aminosäuresubstitutionskarte (die eine Reihe zusätzlicher ETL-Schritte durchlaufen hat) nahtlos aktualisiert.
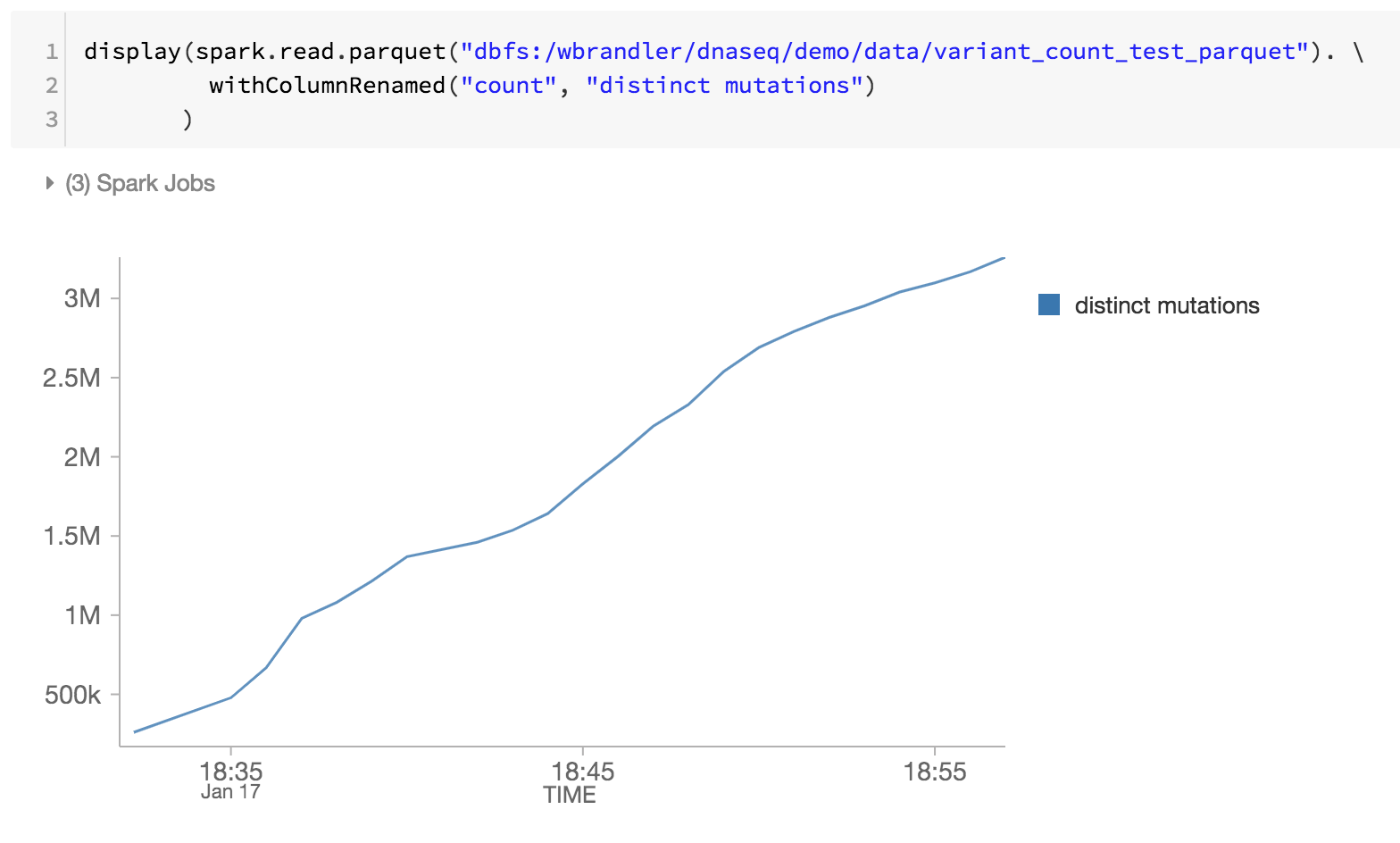
Als letzten Schritt unserer Genomik-Pipeline führen wir auch ein Monitoring der verschiedenen Mutationen durch, indem wir die Databricks Delta Parquet-Dateien in DBFS überprüfen (d. h. die Zunahme der verschiedenen Mutationen im Laufe der Zeit).
Übersicht
Auf der Grundlage der Databricks einheitliche Analyseplattform – mit besonderem Fokus auf Databricks Delta – können Bioinformatiker und Forscher mithilfe der Databricks einheitliche Analyseplattform for Genomik verteilte Analysen mit Transaktionskonsistenz anwenden. Diese Abstraktionen ermöglichen es Datenexperten, Genomik-Pipelines zu vereinfachen. Hier haben wir eine Pipeline zur Qualitätskontrolle genomischer Proben erstellt, die Daten kontinuierlich verarbeitet, sobald neue Proben verarbeitet werden, ohne dass manuelle Eingriffe erforderlich sind. Ganz gleich, ob Sie ETL oder komplexe Analysen durchführen, Ihre Daten fließen schnell und ohne Disruption durch Ihre Genomik-Pipeline. Probieren Sie es noch heute selbst aus, indem Sie das Notebook „Simplifying Genomik Pipelines at Scale with Databricks Delta“ herunterladen.
Erste Schritte mit der Analyse von Genomik im großen Scale:
- Lesen Sie unseren Lösungsleitfadenzu Unified Analytics für Genomik
- Laden Sie das Notebook „Simplifying Genomik Pipelines at Scale with Databricks Delta“herunter
- Registrieren Sie sich für eine kostenlose Testversion von Databricks Unified Analytics for Genomics
Danksagung
Vielen Dank an Yongsheng Huang und Michael Ortega für ihre Beiträge.
Besuchen Sie den Online-Hub von Delta Lake, um mehr zu erfahren, den neuesten Code zu downloaden und der Delta Lake-Community beizutreten.
Erhalten Sie die neuesten Beiträge in Ihrem Posteingang
Abonnieren Sie unseren Blog und erhalten Sie die neuesten Beiträge direkt in Ihren Posteingang.